Charles S. Pavia* a Department of Biomedical Sciences, NYIT College of Osteopathic Medicine, New York Institute of Technology, Old Westbury, NY, United States
b Division of Infectious Diseases, New York Medical College, Valhalla, NY, United States
* Corresponding author: email address: cpavia@nyit.edu
Abstract
The hesitancy and resistance to get vaccinated against COVID-19, by a relatively small but significant part of the general population, has become a serious worldwide problem, and particularly in the United States, despite a vigorous and highly organized governmental advertising campaign promoting vaccination. The unwillingness to get vaccinated has its roots in mostly the spreading of non-scientific, unproven or misleading information. This chapter explains many of the reasons, including an historical connection, behind this anti-vaccine movement, and proposes several possible and feasible remedies to counter this sentiment.
Keywords
COVID-19; SARS-Cov-2; Vaccines; Vaccine hesitancy; Interventions; Pasteur
One evening, as a young teenager in the 1960s, I watched a televised movie entitled “The Story of Louis Pasteur”. It impressed me so much that I began to seriously consider pursuing a career in medicine or one of the biological sciences. I subsequently chose the latter career path and, several years, later I received my Ph.D. degree in microbiology and immunology. The movie was a classic Hollywood production originally released for public viewing in 1937 and was a “Best Picture” nominee. The lead character was the accomplished and well-known actor, Paul Muni, who won a “Best Actor” award from the motion picture industry for his outstanding performance in his portrayal of Pasteur—considered by many in the medical and scientific community to be one of the founding fathers of modern microbiology and immunology. He was a chemist by training (as well as being a self-made accomplished artist), and he coined the term “microbe”, based on his many discoveries and drawings of what he observed under the microscope often to the displeasure of some of his contemporaries, especially the medical establishment. Many of its members were reluctant initially to go along with some of his experimentally derived ideas and concepts, based in part because they seemed quite radical at the time given the limitations of biomedical science 150 + years ago. Despite possibly putting his career and reputation in jeopardy by occasionally going beyond acceptable medical ethics when his work involved certain types of human studies, which were less restrictive then than they are now, Pasteur persevered working tirelessly and with much conviction to prove his sceptics wrong, and indeed he showed through careful experimentation that things (minute living organisms) not visible to the naked eye can indeed cause disease. By doing so, he debunked the previously held belief, by some of his counterparts, in the theory of “spontaneous generation”, and is credited with providing us with the germ theory of disease. Eventually, many of his original sceptics started to accept his ground-breaking findings as being true.
While some parts of the movie may have been slight exaggerations and overly dramatic renditions of actual events of those described in Pasteur's biography (Vallery-Radot, 1929), from which the movie got much of its material, it nonetheless had quite a number of heart-warming and inspiring scenes to go along with scientifically accurate pieces of information. Foremost among Pasteur's many life-altering achievements was his development of two diverse vaccines—one for preventing anthrax in livestock animals, and the other for preventing/treating human rabies (Fig. 1)—both being dreaded diseases having very high levels of mortality, both then and now, in the absence of appropriate medical intervention. With these historical accounts highlighting some of the key events in Pasteur's career as the background, it is amazing and at times disturbing to observe how certain aspects of this past scenario from another era is playing out closely today in the wake of the current COVID-19 pandemic. In this regard, many people, mostly outside the medical community, but a significant few within, have refused or remain hesitant to receive any of the available COVID-19 vaccines. Given the seriousness of COVID-19 with its life-threatening potential and the worldwide emergence of multiple highly contagious/virulent mutants, such as the Delta variant, of the etiologic agent SARS-CoV-2, this reluctance to get vaccinated is very difficult to understand and has led to tragic results. In addition, the vaccine options against COVID-19 are multiple (Table 1) and involve either the use of a purified mRNA component of the virus or a viral vector formulation, with both types having excellent safety and efficacy results based on extensive clinical trials and, as of September 2021, have been approved or are nearing full approval from the FDA for use in the United States. In July 2021, as part of a nationally televised briefing (White House COVID-19 Response Team, 2021), highly regarded public health officials from the U.S. Centers for Disease Control and Prevention (CDC) and the National Institutes of Health provided updates on a new surge nationwide in hospitalizations and deaths due to COVID-19, and have pointed out correctly that we are now entering into a “pandemic of the unvaccinated”. This comment by the director of the CDC is supported by data showing that in some locations > 99% of the current wave of victims developing serious disease are among the unvaccinated, leading to the obvious conclusion that these negative outcomes could have been avoided if these victims had chosen to receive the vaccine beforehand. This surge in cases has continued and even increased in many locations in the months that followed.
 Fig. 1 This relatively large and impressive portrait of Pasteur, done by Albert Edelfeldt in 1885 and having the dimensions of 155 cm in height and 127.5 cm in width, shows him observing one of his rabies “cultures” using rabbit spinal cord material that presumably enabled him to grow the rabies virus. The original painting hangs majestically in the dining room of Pasteur's former home/laboratory located on the campus of the Pasteur Institute in Paris.
Fig. 1 This relatively large and impressive portrait of Pasteur, done by Albert Edelfeldt in 1885 and having the dimensions of 155 cm in height and 127.5 cm in width, shows him observing one of his rabies “cultures” using rabbit spinal cord material that presumably enabled him to grow the rabies virus. The original painting hangs majestically in the dining room of Pasteur's former home/laboratory located on the campus of the Pasteur Institute in Paris.
Table 1
|
Comparison of the key features of the COVID-19 vaccines that are produced by the four leading manufacturers. |
|||||||
|
Vaccine manufacturer |
Type of vaccine |
Number of doses |
Storage method |
Efficacy |
Authorized for use in the U.S.a |
EUAb pending final approval |
Serious adverse events |
|
Moderna |
mRNA |
2c |
4–10 oC |
> 94% |
Yes |
Yesd |
Raree |
|
Pfizer |
mRNA |
2c |
− 80 oC |
> 95% |
Yes |
Yesd |
Raree |
|
Janssen |
Viral vector |
1c |
4–10 oC |
66.3% |
Yes |
Yes |
Yesf |
|
Astra-Zeneca |
Viral vector |
2 |
4–10 oC |
63%–84% |
Yes |
Yesd |
Yesf |
a Information that was available as of August 2021.
b In the U.S., an EUA (Emergency Use Authorization) was originally given for these vaccines for people aged > 16, which was subsequently expanded to > 12 years of age.
c As of October 2021, additional booster injections were highly recommended for these vaccines by the FDA and CDC.
d In the U.K., an EUA no longer applies for the Pfizer, Moderna and Astra-Zeneca vaccines which now have been granted final approval for use. In the U.S. as of August 2021, the Pfizer vaccine was given FDA approval, while the other 3 are undergoing further evaluation for full approval by the FDA.
e Milder and temporary forms of myocarditis and pericarditis have occurred in a small number of young adults with these vaccines.
f Blood clots have been reported in a small number of female vaccine recipients < 50 years of age with these vaccines.
What are the reasons for such hesitancy/resistance or for some of the obstacles to get vaccinated? Here are some major examples (summarized in Table 2):
Table 2
|
Characteristics of the anti-COVID vaccine phenomenon. |
|
|
Reason |
Basis or source |
|
Delay in getting vaccinated |
Vaccine must be fully approved |
|
Fear of adverse events |
Some serious side effects have been reported |
|
Anxiety/fear of inoculations |
Behavioural/psychological reaction |
|
Inaccessibility to vaccine sites |
Problem with logistics or infrastructure |
|
Freedom of choice/expression |
Political convictions or posture |
|
Moral or religious objections |
Misguided religious teachings or upbringing |
|
Vaccine is not safe or effective |
Misinformation/conspiracy theories spread mostly by disreputable, non-scientific/non-medical people |
|
Experimental data is fake |
|
It is important to realize that the anti-vaccine sentiment is not limited to the United States, but it is a global problem (Hornsey, Harris, & Fielding, 2018) and had been well established as a recurring theme multiple times prior to the onset of the current pandemic often with devastating consequences. Even certain political leaders and other professionals of varying degrees of influence have joined the anti-vaccine bandwagon. This is reminiscent of what has occurred in the pre-COVID days with certain other infectious diseases (reviewed in Smith, 2017), where “herd immunity” which can develop when a sufficient number of people survive a natural infection, has been invoked as the only mechanism whereby the spread of SARS-Cov-2 can be blocked leading allegedly to its eventual disappearance within the general population. Evidence in support of the pervasiveness and effects of these untenable reasons against the COVID-19 vaccine comes from recently published data put forth by the Kaiser Foundation (Hamel, Kirzinger, Munana, & Brodie, 2021), and these are as follows.
In terms of demographics, 27% of the people in the United States remains vaccine-resistant, saying they probably or definitely would not get a COVID-19 vaccine even if it were available for free and deemed safe by scientists and public health officials. Vaccine hesitancy is highest among those who identify as belonging to a certain political party (42%), those in the 30–49 age group (36%), and rural residents (35%), especially those living in certain parts of the Midwest and Southeast sections of the United States where less than 50% of the population has gotten vaccinated, and where concurrently there has been an alarming new surge (as of July/August 2021) in COVID-19 cases requiring hospitalization. In addition, 35% of African American adults (a group that has had to bear a disproportionate burden of the effects of the pandemic) say they definitely or probably would not get vaccinated, citing as major reasons that they don't trust vaccines in general (47%) or that they are worried they may get sick with COVID-19 from just the vaccine alone (50%). This situation suggests that messages combatting particular types of misinformation may be especially important for increasing vaccine confidence among this group. Perhaps most astonishingly, are the data showing that as much as one third of those who say they are considered to be essential workers and 29% of those who perform services in a health care delivery setting will not take the vaccine. The resistance of people to receive COVID-19 vaccines is even more perplexing given the excellent track record of other vaccines, some of which have been with us for nearly 100 years as part of routine preventive care. These have been designed to protect us against smallpox, tuberculosis (with BCG) and polio, followed, more recently, with the DPT (diphtheria, pertussis, tetanus), MMR (measles, mumps, rubella), and chickenpox vaccines. It should be noted that, in most jurisdictions, the latter three childhood vaccine combinations are required before young children can start school at the elementary level.
So, in light of the foregoing, how would a modern-day Pasteur react to the relatively high level of resistance to get vaccinated against COVID-19, and what might be a solution to this problem? Given his personality for scientific rigour combined with the overwhelming evidence showing the vaccine's safety (with few exceptions) and efficacy, he probably would be highly disappointed with the overall public response to getting vaccinated, especially by those who remain unconvinced on why the vaccine is so important from a public health perspective, and why there is such lack of confidence given the many advances made in medicine over the past 100 + years. He would also be very outspoken and adamant against the sceptics who perpetuate false or misleading information about the vaccine. If requested (and this would seem highly likely given his prestige), he would probably be making frequent public appearances on various news programs similar to what the current wave of public health medical experts are doing now. He might even have his own podcast where he could dispense valuable information to the misinformed or uninformed public and reinforce the importance of getting vaccinated. He would probably find it amusing and perhaps a bit misguided that incentives, such as monetary and other rewards, are being offered in certain parts of the United States to try to get people vaccinated, even though vaccines are being administered free of charge in most, if not all, locations. To him, it would seem like deja vu in terms of what he experienced with the obstacles that he had to face in dealing with his disbelieving scientific and medical contemporaries, when he was making his groundbreaking and life-changing discoveries in the late 1800s, at a time when complex biomedical processes were still poorly understood, and initially underappreciated.
Another variable that awaits more clarity is whether and/or when patients, who may have acquired some form of natural immunity after recovering from COVID-19, should get vaccinated, if at all. This provokes the following question: are they no longer susceptible, or are less vulnerable, to serious disease, after re-exposure to SARS-CoV-2, that getting vaccinated would be unnecessary? It is believed, however, that solid immunity against COVID-19 may gradually wane (Centers for Disease Control and Prevention, 2021a), and thus booster injections with one of the available vaccines may still be necessary in order to provide optimal protection against re-infection. A somewhat related recommendation, also coming from the CDC, states that, starting in September 2021, another or third booster shot will have to be given to earlier vaccines to maximize vaccine-induced protection and prolong its durability, and that these booster shots will be offered for all Americans, who had received their last (second) dose or had recovered from a prior infection with SARS-CoV-2 at least 8 months previously.
Beyond what a contemporary Pasteur might be inclined to do, what else could be done? Various organizations have made numerous suggestions, along these lines, including several promoted and offered by the CDC (Centers for Disease Control and Prevention, 2021b), and a few more that this author will offer. They include the following services that could be implemented. A large number of healthcare professionals and qualified scientists (primarily microbiologists and immunologists) are needed to support COVID-19 vaccination efforts nationwide. It is important they receive the necessary training to effectively meet the demands of their roles. Training must be ongoing as new COVID-19 vaccines become available and as vaccine recommendations evolve and more is learned about the vaccines and how to improve and maintain the vaccination process. In terms of educating the lay public, they are essential to ensuring that the American population is vaccinated safely as soon as possible, based on a true understanding on why this is an important undertaking. Furthermore, as parents' most trusted source of information on vaccines, paediatric healthcare professionals play a critical role in helping parents and guardians understand the importance of COVID-19 vaccination and assuring them that COVID-19 vaccines are safe and effective, and that they are important steps in protecting their children's health (both physical and mental). Parents need to be reminded that fully vaccinated people are less likely to spread the virus that causes COVID-19. Getting all family members 12 years and older (and when recommended, children less than 12 years of age) vaccinated can protect other family members around you, including people at increased risk for severe illness from COVID-19. Students also need to be reminded that, after they are fully vaccinated, they will be able to resume many activities with family and friends, such as going to parties, weddings, graduation exercises, and other social gatherings that they have missed due to prior restrictions that were imposed on everyone at the height of the pandemic.
As part of the education process, students should be contacted and encouraged to learn more from reliable sources derived from rigorously peer-reviewed articles, especially those found on medically-based Internet sites, such as PubMed, reputable blog and social media posts, properly mentored student-driven publications and social groups. A feedback mechanism should be created in order for students to ask questions and get a meaningful response quickly about COVID-19 vaccination, such as by using either e-mail, online video conferences (via Zoom, Skype, Facetime or any other similar provider), or by phone number. Any student concerns or questions should be proactively addressed and the spread and harm of misinformation should be countered by sharing credible and accurate information. Students, as well as other participants, should be warned about relying on unregulated or non-scientifically based sources of information that are circulating on the Internet or other similar platforms. Students should be warned about the dangers caused by misinformation and disinformation, and health literacy should be promoted as a means to be fully informed in understanding the benefits of being vaccinated as well as the negative outcomes that could arise if the vaccine is not received.
Although much of the preceding suggestions pertain mostly to the adolescent age group, similar interventions should also be considered for those attending colleges and universities after completing high school, to reinforce and update what students had learned previously about vaccines. Hopefully, as this younger and now well-informed generation matures into adulthood, they will be representative of a population having much greater acceptance and less resistance to getting vaccinated when deemed necessary by experts in the health care community for both now and, just as important, later on, when other future serious outbreaks may arise.
As an additional approach, schools, at both the elementary and high school level, can also take the initiative by recruiting the suitably trained medical professionals or scientists to come to their classrooms, when this becomes allowable, and supplement the curriculum by providing a better understanding of the vaccine process. In order to accomplish this task more effectively, local health departments, especially those within the jurisdiction of the schools, should provide the schools with a registry of trained personnel who would be willing to come to the schools and share their knowledge and expertise pertaining to the COVID-19 vaccines with the students, along with the teachers and administrators, as well as answering any questions or concerns that students may have on this topic, preferably at no additional cost to the school district.
Another possible program worth considering would be for medical schools to offer a short course on the vaccine process to be attended by elementary and high school teachers to better educate them on this topic with all of its subtle nuances. Upon completion of the course, the teachers would receive a certificate of recognition (similar to CME credits) showing that they had participated successfully in this learning exercise. As such, they would be well equipped to return to their respective schools with what they had learned about vaccines and share this information with their students, during one of their standard classroom sessions or remotely (typically when the subject of “Biology” is being taught or during “Health Class”). Presumably, the best time to give this course would be during the summertime when most medical schools are less active with their didactic responsibilities as part of the regular pre-clinical curriculum, and where most of the first- and second-year medical students are not on campus due to being on an extended break for their summer vacation. In so doing, this would not impose an undue burden on the school's infrastructure or their faculty who would be providing this pertinent information to the participants in this program.
In conclusion, people should not be fearful of receiving any of the available anti-COVID-19 vaccines. The benefits of getting jabbed far outweigh the minimal risks of having an adverse or serious outcome. Historically, vaccines have had an excellent track record in terms of saving lives, reducing morbidity caused by a wide variety of infectious agents, and easing the burden of the health care community and infrastructure. In addition, the various and somewhat innovative interventions put forth in this chapter, that are designed to educate people about the COVID-19 vaccines, with the goal of getting as many people vaccinated, especially towards dealing with the existing anti-vaccine trend, may be seen as a daunting task. Accordingly, it will require a well-organized and coordinated effort, a well-equipped infrastructure and cooperative interactions among all parties involved in order to make these interventions a success and not just wishful thinking. Such opportunities to try to gain much wider vaccine acceptance would perhaps be in keeping with the often-quoted comment by the legendary Pasteur of: “dans les champs de l'observation, le hasard ne favorise que les esprits préparés” (“in the field of observation, chance favours only the prepared mind”) (Pasteur, 1939), in honour of him as we approach in the coming year of 2022 the bicentennial of his birth.
References
Centers for Disease Control and Prevention, 2021a Centers for Disease Control and Prevention. Frequently asked questions about COVID-19 vaccination. 19 August 2021 https://www.cdc.gov/coronavirus/2019-ncov/vaccines/faq.html#:~:text=Yes%2C%20you%20should%20be%20vaccinated,ve%20already%20had%20COVID%2D19. 2021a.
Centers for Disease Control and Prevention, 2021b Centers for Disease Control and Prevention. How schools can support COVID-19 vaccination. June 29, 2021 https://www.cdc.gov/vaccines/covid-19/planning/school-located-clinics/how-schools-can-support.html. 2021b.
Hamel, Kirzinger, Munana and Brodie, 2021 Hamel L., Kirzinger A., Munana C., Brodie M. KFF COVID-19 monitor: December 2020.https://www.kff.org/coronavirus-covid-19/report/kff-covid-19-vaccine-monitor-december-2020/. 2021 accessed on July 22, 2021.
Hornsey, Harris and Fielding, 2018 Hornsey M.J., Harris E.A., Fielding K.S. The psychological roots of anti-vaccination attitudes: A 24-nation investigation. Health Psychology. 2018;37(4):307–315. doi:10.1037/hea0000586.
Pasteur, 1939 Pasteur L. Discours prononcé à Douai, le 7 décembre 1854, à l'occasion de l'installation solennelle de la Faculté des lettres de Douai et de la Faculté des sciences de Lille. In: Vallery-Radot P., ed. Speech delivered at Douai on December 7, 1854 on the occasion of his formal inauguration to the Faculty of Letters of Douai and the Faculty of Sciences of Lille. Oeuvres de Pasteur; Paris, France: Masson and Co; 131. 1939;Vol. 7 http://gallica.bnf.fr/ar k:/12148/bpt6k7363q/f137.chemindefer.
Smith, 2017 Smith T.C. Vaccine rejection and hesitancy: A review and call to action. Open Forum Infectious Diseases. 2017. ;4(3). https://doi.org/10.1093/ofid/ofx146.
Vallery-Radot, 1929 Vallery-Radot R. The life of Pasteur. Garden City, NY: Garden City Publishing Co., Inc; 1929.
White House COVID-19 Response Team, 2021 White House COVID-19 Response Team. White House COVID-19 Response Team Briefing.https://www.c-span.org/video/?513463-1/cdc-director-warns-pandemic-unvaccinated. 2021 July 16, 2021.
Chapter 8: The emergence of SARS-CoV-2 variants of concern in Australia by haplotype coalescence reveals a continental link to COVID-19 seasonality
Tre Tomaszewskia; Volker Gurtlerb; Kelsey Caetano-Anollésc; Gustavo Caetano-Anollésa,* a Evolutionary Bioinformatics Laboratory, Department of Crop Sciences, University of Illinois, Urbana, IL, United States
b RMIT University, Melbourne, VIC, Australia
c Callout Biotech, Albuquerque, NM, United States
* Corresponding author: email address: gca@illinois.edu
Abstract
SARS-CoV-2 continues to evolve, even after implementation of public-wide vaccination, as can be observed by an increasing number of mutations over time. Compared to responses by the United States and European countries, the disease mitigation strategies employed by the Australian government have been swift and effective. This provides a unique opportunity to study the emergence of variants of concern (VOCs) at many latitude levels in a country that has been able to control infection for the majority of the pandemic. In the present study, we explored the occurrence and accumulation of major mutations typical of VOCs in different regions of Australia and the effects that latitude has on the establishment of VOC-induced disease. We also studied the constellation of mutations characteristic of VOCs to determine if the mutation sets acted as haplotypes. Our goal was to explore processes behind the emergence of VOCs as the viral disease progresses towards becoming endemic. Most reported COVID-19 cases were in largest cities located within a –30°S to − 50°S latitude corridor previously identified to be associated with seasonal behavior. Accumulation plots of individual amino acid variants of major VOCs showed that the first major haplotypes reported worldwide were also present in Australia. A classification of accumulation plots revealed the existence of 18 additional haplotypes associated with VOCs alpha, delta and omicron. Core mutant constellations for these VOCs and curve overlaps for variants in each set of haplotypes demonstrated significant decoupling patterns, suggesting processes of emergence. Finally, construction of a “haplotype network” that describes the viral population landscape of Australia throughout the COVID-19 pandemic revealed significant and unanticipated seasonal patterns of emergence and diversification. These results provide a unique window into our evolutionary understanding of a human pathogen of great significance. They may guide future research into mitigation and prediction strategies for future VOCs.
Keywords
Amino acid substitution; Epidemic calendar; Genetic diversity; Haplotype; N-terminal domain; Mutation; Proteome; SARS-CoV-2; Seasonality; Variants of concern; Virus
1: Introduction
The COVID-19 pandemic illustrates how a virus is capable of overcoming barriers to its persistence by rapidly changing its genomic makeup. Thanks to extensive worldwide genome sequencing efforts, researchers now have direct access to information about the levels of genetic variation unfolding in the evolving viral population, as well as variations associated with physiological responses of human or animal hosts. As of January 15, 2022, the GISAID initiative (https://www.gisaid.org) sponsored by many governments in partnership with public health and research institutions (Elbe & Buckland-Merrett, 2017; Khare et al., 2021; Shu & McCauley, 2017) has collected over 7 million genomic sequences of the SARS-CoV-2 virus, making them freely accessible to the scientific community for analysis. In parallel, the open-source Nextstrain project (https://nextstrain.org) made available a continuously updated phylogenomic view of this data alongside with powerful and portable analysis and visualization tools. These resources provide a unique window into our evolutionary understanding of a human pathogen of great significance.
Genetic variation refers to the existence of differences among the genomes of a set of closely or more distantly related organisms or viruses. This diversity in genomic makeup constitutes one primary source of phenotypic diversity, i.e., diversity in observable biological characteristics (traits). The other primary source involves epigenetic variation. Genetic variation results from the effects of a multiplicity of processes, including spontaneous mutation, error-prone replication, recombination, and genetic exchange. Mutations can be small-scale or large-scale alterations in the nucleotide sequence of genomic DNA present in most life forms or RNA typical of some viruses. Small-scale alterations include exchange (substitution), addition (insertion) or removal (deletion) of nucleotides in a sequence. Large-scale alterations include duplications, translocations or inversions of larger nucleic acid segments. Regardless of their nature, the physiological impact of these alterations typically materializes at the level of proteins or functional RNA. Nucleotide triplets for the most part encode for amino acids, which control the physiological activities of the cell by serving as the building blocks of proteins. A non-synonymous mutation leading to change in one or more amino acids of a polypeptide sequence can alter the structure and functioning of the mutated protein. For example, in the case of the SARS-CoV-2 virus, a single amino acid substitution at position 614 of the viral spike glycoprotein (from aspartic acid to glycine, referred to as the D614G mutation) that occurred during the first wave of the pandemic resulted in increased viral transmissibility (Voltz et al., 2021). Distinct viruses holding one or a unique constellation of these types of mutations are generally called “variants” (Lauring & Hodcroft, 2021). At the protein level, mutations cause amino acid substitutions (e.g., D614G), which are called “amino acid variants.”
Genetic variants arise in the context of evolving populations. Thus, mutations in single or multiple genomic locations are often the subject of evolutionary effects on fitness (e.g., natural selection) or the effect of chance events on sampling (e.g., genetic drift). In the case of viruses, their fate can depend for example on whether they confer competitive advantage to viral replication, rates of transmission, immune escape, or virulence. Mutations that do not provide an advantage are often eliminated from the population, unless “founder effects” on newly established viral populations extend their persistence. Epidemiologically, a mutation that alters transmissibility, disease severity, or immune or vaccine escape becomes a “mutation of concern” (MOC) and its presence in a variant a candidate for surveillance and response. More importantly, a “variant of concern” (VOC) is a variant of the virus exhibiting a constellation of mutations associated with statistically significant and experimentally verified increases in virus transmissibility, disease severity, immune and vaccine escape, diagnostic test evasion, or other clinical or epidemiological criteria of significance. VOCs become immediate priority for surveillance and response, especially when their prevalence increases worldwide.
Haplotypes are sets of mutations that are often inherited together. In the case of viruses, haplotypes are known to represent mutations that appear tightly linked with each other. For example, the D614G mutation of the SARS-CoV-2 spike protein is part of an haplotype of four mutations that also alter the NSP12 polymerase (P323L), 5′ untranslated region (UTR), and silently the NSP3 papain-like protein (F106F). This haplotype was the first gene set to be fixed in the worldwide viral population during the first wave of the COVID-19 pandemic in early 2020. Since VOCs are mutation constellations reflecting successful viral variants that have overtaken the global population, there is an implicit assumption that these constellations are stable haplotypes. This assumption however has not been fully tested. Here we explore the appearance and accumulation of major mutations typical of VOCs in Australia as the viral disease progresses towards becoming endemic. We study the constellation of mutations characteristic of VOCs to determine if the mutation sets acted as haplotypes and to test if these haplotypes are the subject of regional variation in Australia. Our goal is to explore processes behind the emergence of VOCs in a viral pandemic, including effects of viral seasonal behavior.
2: Methods
The metadata for 7,175,152 SARS-CoV-2 genome sequences was downloaded from GISAID (https://www.gisaid.org) on January 18, 2022. The metadata were then filtered for sequences marked “complete” with “human” hosts and a “location” field containing the case-insensitive term “Australia,” which reduced the set to 58,378 sequences. Of these, the sequences were collected between January 1, 2020 and January 13, 2022 (743 days) and submitted for deposition to GISAID between January 31, 2020 and January 17, 2022 (717 days) (see acknowledgements in Supplementary information for complete list of Accession IDs used).
The Australian region (state/territory) for each sequence was then extracted from the “location” field, resulting in sequences belonging to each of eight regions (Table 1). The metadata was then labeled by “period,” which was derived from the collection date's year and calendar quarter. This was done so that, for example, January 1, 2020 to March 31, 2020 was designated as “Period 1” and January 1, 2022 to March 31, 2022 as “Period 9”.
Table 1
|
Sequences by state/territory of Australia. |
||
|
State/territory |
Number of sequences |
Proportion of sequences |
|
Victoria |
22,011 |
0.434 |
|
New South Wales |
21,922 |
0.432 |
|
Queensland |
3142 |
0.062 |
|
Australian Capital Territory |
1927 |
0.038 |
|
South Australia |
761 |
0.015 |
|
Western Australia |
588 |
0.012 |
|
Tasmania |
222 |
0.004 |
|
Northern Territory |
171 |
0.003 |
The sequence metadata provide the field “AA Substitutions,” which contains a comma-separated list of each identified amino acid substitution (against the reference sequence NC_045512) by protein name, reference amino acid, amino acid location within the sequence, and the substituting amino acid (formatted as < Protein Name >_). The list for each sequence was transformed into a one-hot encoding for each of the 9281 mutational substitutions, indexed by the Accession ID, and the derived region and period.
Grouped by these derived attributes, a simple summation of each possible mutation across sequences provided the occurrence count for each region-period grouping. Dividing the summation of any mutation by the total number of sequences within the group provided the prevalence of each amino acid substitution for each region-period. These groups were then aggregated and regrouped by mutation, enabling regional comparisons of the prevalence of each mutation by time period (year-quarter).
Since substitution groups containing low variance or spurious substitutions were undesired for further analysis, the groups were filtered by a “relevancy” heuristic. The prevalence of any given substitution was required to be above a threshold of 0.1 in more than 2 regions for at least one period, although there was no requirement that this threshold was met during the same period.
Review of the initial results revealed certain variant-specific substitutions occurring in improbable time-periods (e.g., simultaneous mutations appearing in significant amounts 3 quarters prior to announced detection). Further analysis of the data indicated 292 instances were labeled with collection dates that were only identified by year. These were reconciled by using the submission date as a proxy. The entire process detailed above was then repeated to achieve final results.
Extraction and transformation was performed using the Python library Pandas (McKinney, 2010; Pandas Development Team, 2022). The Python library “matplotlib” (Caswell, Droettboom, Lee, et al., 2021; Hunter, 2007) was used to produce raw plots of the data, followed by an additional arrangement, annotation, and graphical modification using Adobe Illustrator. Source code and supplementary information can be found at https://doi.org/10.1016/bs.mim.2022.03.003.
3: VOCs in Australia
SARS-CoV-2 variants are organized around a master genomic sequence of the virus that originated in the city of Wuhan in China (accession NC_045512.2, version March 30, 2020; previously “Wuhan seafood market pneumonia virus”). Many mutations have been added to, and subtracted from, this master sequence since the beginning of the pandemic. These genomic changes can be traced through their phylogenies (Fig. 1A). Phylogenies are hypotheses of history and genealogical relationship among groups of genomes (evolving taxa) in the form of tree structures (networks without reticulations). They harbor specific connotations of ancestry and an implied time axis, which enables the study of important epidemiological phenomena such as viral spread, variant introduction, and rates of genomic change and epidemic growth. Splits in the branches of these trees define clades, i.e., groups of taxa with a common evolutionary origin. Clades are often defined by the statistical distribution of distances between phylogenetic clusters followed by lineage merging based on mutations that are shared. As of February 2022, genome sequences have been clustered into 11 GISAID clades (L, S, O, V, G, GH, GR, GV, GRY, GK, and GRA) or 23 Nextstrain clades defined by a year-letter nomenclature. In the case of Nextrain clades, a new clade must differ by at least 2 mutations from its parent major clade (Hodcroft, Hadfield, Neher, & Bedford, 2020). The tree shown in Fig. 1A uses the Nextstrain nomenclature to pinpoint the evolutionary appearance of major VOCs along a timeline that originated when the first two clades (19A and 19B) diverged from each other. In the figure, the current VOC omicron wave is represented by major clades 21 K and 21 L, which originated from a larger more basal clade that gave rise to VOC alpha. Nextstrain clades of VOC omicron correspond to the recent GISAID clade GRA. In addition, clades can be defined at lower granularity using the Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin) tool that automatically assigns sequences to lineages and sublineages (Rambault et al., 2020). For example, VOC omicron corresponds to Pangolin lineage B.1.1.529 and the previously prevalent VOC delta to lineage B.1.617.2, both of which harbor numerous sublineages.
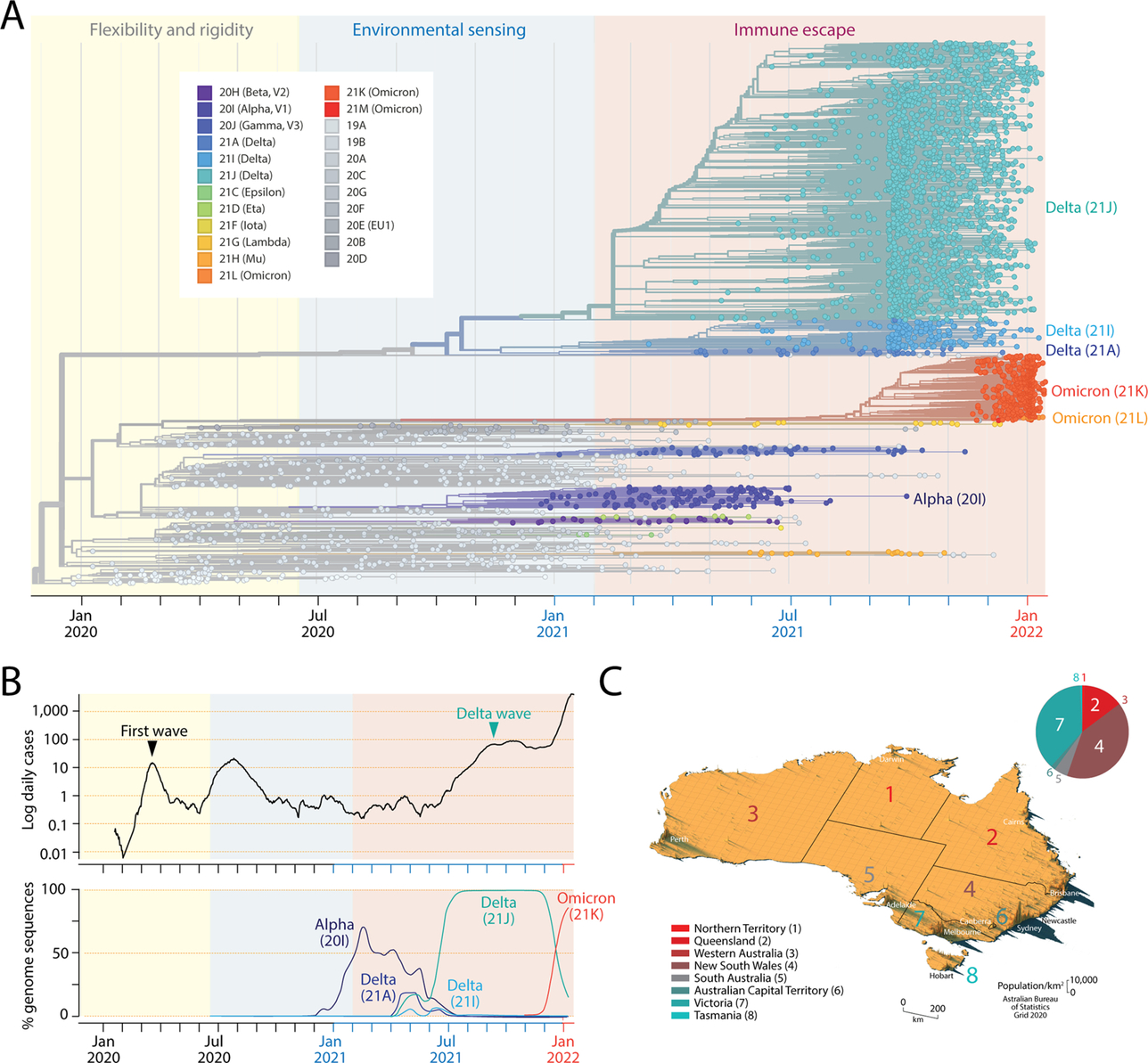 Fig. 1 The mutational landscape of the SARS-CoV-2 virus at the beginning of 2022 and its historical spread throughout the Australian continent. (A). A maximum likelihood phylogenetic tree describes the worldwide history of the SARS-CoV-2 genome. The timetree of 3347 genomes randomly sampled between December 2019 and January 2022 was obtained from Nextstrain (https://nextstrain.org) on January 15, 2022. The tree unfolds time of genome collection date from left to right. Its leaves (taxa indicated with circles) are colored according to clade (group of taxa with a common evolutionary origin) and emerging variants of concern (VOCs) nomenclature. The origin of VOCs occurs when a clade originates along branches of the phylogeny. Note the early arrival of VOC alpha, followed by VOC delta and then VOC omicron. The timeline of clades and VOCs show three successive phases driven by proteome flexibility and rigidity, environmental sensing and vaccine-driven immune escape, which are shaded in light yellow, blue and salmon, respectively (Caetano-Anollés, Hernandez, Mughal, Tomaszewski, & Caetano-Anollés, 2022). (B). Plots show numbers of newly confirmed cases per 1000 people (in logarithmic scale and as 7-day rolling averages) and smooth percentages of genomes holding major VOCs in Australia since the beginning of the recorded COVID-19 pandemic. COVID-19 and genome data are derived from Johns Hopkins Univ., CSSE and GISAID, respectively. (C). Spike map showing the population density of Australia as a grid of vertical bars depicting number of people per square kilometer of land area (courtesy of Alasdair Rae, Automatic Knowledge Ltd., Sheffield, UK). The different states/territories of Australia are identified with colored numbers using shades that correspond to increasing latitudes of their population medians across cells (from red to turquoise). The pie chart describes the relative number of total cases (cumulative, confirmed and under investigation) reported by the Department of Health, States and Territories for individual regions on February 4, 2022.
Fig. 1 The mutational landscape of the SARS-CoV-2 virus at the beginning of 2022 and its historical spread throughout the Australian continent. (A). A maximum likelihood phylogenetic tree describes the worldwide history of the SARS-CoV-2 genome. The timetree of 3347 genomes randomly sampled between December 2019 and January 2022 was obtained from Nextstrain (https://nextstrain.org) on January 15, 2022. The tree unfolds time of genome collection date from left to right. Its leaves (taxa indicated with circles) are colored according to clade (group of taxa with a common evolutionary origin) and emerging variants of concern (VOCs) nomenclature. The origin of VOCs occurs when a clade originates along branches of the phylogeny. Note the early arrival of VOC alpha, followed by VOC delta and then VOC omicron. The timeline of clades and VOCs show three successive phases driven by proteome flexibility and rigidity, environmental sensing and vaccine-driven immune escape, which are shaded in light yellow, blue and salmon, respectively (Caetano-Anollés, Hernandez, Mughal, Tomaszewski, & Caetano-Anollés, 2022). (B). Plots show numbers of newly confirmed cases per 1000 people (in logarithmic scale and as 7-day rolling averages) and smooth percentages of genomes holding major VOCs in Australia since the beginning of the recorded COVID-19 pandemic. COVID-19 and genome data are derived from Johns Hopkins Univ., CSSE and GISAID, respectively. (C). Spike map showing the population density of Australia as a grid of vertical bars depicting number of people per square kilometer of land area (courtesy of Alasdair Rae, Automatic Knowledge Ltd., Sheffield, UK). The different states/territories of Australia are identified with colored numbers using shades that correspond to increasing latitudes of their population medians across cells (from red to turquoise). The pie chart describes the relative number of total cases (cumulative, confirmed and under investigation) reported by the Department of Health, States and Territories for individual regions on February 4, 2022.
VOCs emerged in October 2020, less than half a year after the first wave of the pandemic. VOC alpha (also known as Nextstrain clade 20I or Pangolin lineage B.1.1.7) appeared in the United Kingdom and was the first to expand quickly worldwide, probably correlated with significant increases in transmissibility and infection rates (Davies et al., 2021). VOC beta (20H, B.1.351) appeared in December 2020, following its first report in South Africa, and VOC gamma (20 J, P.1) appeared in the Amazonian region of Brazil in January 2021. The highly prevalent VOC delta (21A, B.1.617.2), while first discovered in India in October 2020, became predominant worldwide in June 2021, almost completely replacing other developing VOCs. Finally, VOC omicron was first identified in Botswana and South Africa early in November 2021 and is currently sweeping the world, replacing VOC delta. A global analysis of the spread of the different VOCs (except omicron) and an estimate of effective reproduction numbers revealed rapid replacement of previously circulating variants and transmissibility increases ranging from 25% (alpha) to 97% (delta) (Campbell et al., 2021). These estimates are expected to increase substantially with VOC omicron.
We here focus on the COVID-19 pandemic in Australia and the effects that latitude has on the establishment of VOC-induced disease. Compared to responses from the US and European countries, the disease mitigation strategies employed by federation and local governments of the Australian Commonwealth have been swift and effective. This provides a unique opportunity to study VOC emergence at many latitude levels in a country that has been able to control infection for the majority of the pandemic (Fig. 1B). The first confirmed case of COVID-19 was identified in Victoria on January 25, 2020. Both the central government and individual states responded swiftly to the outbreak by closing borders. This controlled the first wave to some degree by the beginning of April. However, a second and more deadly wave emerged in Victoria during May and June 2020. Although it was largely localized to Melbourne, it was considerably more widespread than the initial wave. Strict lockdown managed to control the disease by November 2020. In order to curb cluster outbreaks, Australia pursued a zero-COVID public health policy of suppression (i.e., “find, test, trace, isolate and support”) that minimized domestic community transmission, enforced strict international border controls, and curbed local outbreaks via lockdowns and exhaustive contact tracing. The policy lasted until late 2021. Despite efforts and a nationwide vaccination program, VOC delta levels increased in April 2021 and a “delta wave” overtook the country in June 2021 with a significant outbreak in New South Wales. Major city lockdowns during July through December 2021 were unable to suppress the rise of case numbers, which was notably exacerbated by the VOC omicron wave that began in 2022. At the beginning of December 2021 there were 211,654 reported cases. After only 2 months (up to February 4, 2022), that number of total cases increased to 2,319,029, with 940,596 cases corresponding to New South Wales and 870,416 to Victoria. Despite these large case numbers, only 4073 total deaths were reported for the entire country. We note that the “delta wave” that started in April 2021 and was predominant for a period of 5–6 months was largely responsible for a significant number of these deaths. The first cases of VOC omicron were reported in Sydney on November 28, 2021, and in Darwin and Sydney on November 29, 2021, all infected travelers returning from southern Africa. Fig. 1B describes the percentage of sequenced genomes corresponding to the main VOCs alpha, delta and omicron that were present in Australia. Remarkably, VOC omicron (21K) took over the identity of most of genome samplings in less than a month, replacing the fully prevalent VOC delta (21J).
Australia is inhabited by 26 million people, making the country the most populous in Oceania. However, because of its significant size (the 6th largest nation in the world), Australia has a very low population density of 3 people/km2. Furthermore, most people live in major urban areas, which largely correspond to the capital cities of the state/territories. The largest cities include Sydney (~ 4.6 million in habitants), Melbourne (~ 4.2 million), Brisbane (~ 2.2 million), Perth (~ 1.9 million) and Adelaide (~ 1.2 million). The Gold Coast, Newcastle and Wollongong add an extra ~ 1.2 million. Fig. 1C shows a spike map of the population density of Australia. It identifies the different states/territories of Australia with numbers colored according to the latitudes of their population medians: 1, Northern Territory (− 12°S); 2, Queensland (− 27°S); 3, Western Australia (− 32°S); 4, New South Wales (− 33°S); 5, South Australia (− 35°S); 6, Australian Capital Territory (ACT) (− 35.2°S); 7, Victoria (− 39°S); and 8, Tasmania (− 43°S). Regions 1–4 can be dissected from regions 5–8 by a –34°S latitude transect, separating the largest cities of Sydney and Melbourne from each other by 4°, 713 km air distance, and a state boundary half-way between the two cities. Most reported COVID-19 cases correspond to the largest cities located within a − 30°S to − 50°S latitude corridor, which was previously identified to be associated with seasonality during the first wave of the pandemic (Caetano-Anollés et al., 2022). A pie chart describing the proportion of total cumulative cases in states/territories shows that 88% of cases appeared in New South Wales and Victoria, driven mainly by the Sydney and Melbourne metropolitan areas (Fig. 1C). A comparison of lineages identified in genome sequences sampled from these two states before the rise of VOC omicron on October 18, 2021 showed Pangolin sublineage B.1.617.2.30 was almost exclusively observed in Victoria following a survey of 4184 and 13,536 sequences from New South Wales and Victoria, respectively (Fig. 2). Other sublineages were also differentially present in the two states. These initial results suggest a seasonal underpinning of the genetic differences responsible for lineage diversification. This initial exploration prompted us to undertake a more exhaustive analysis.
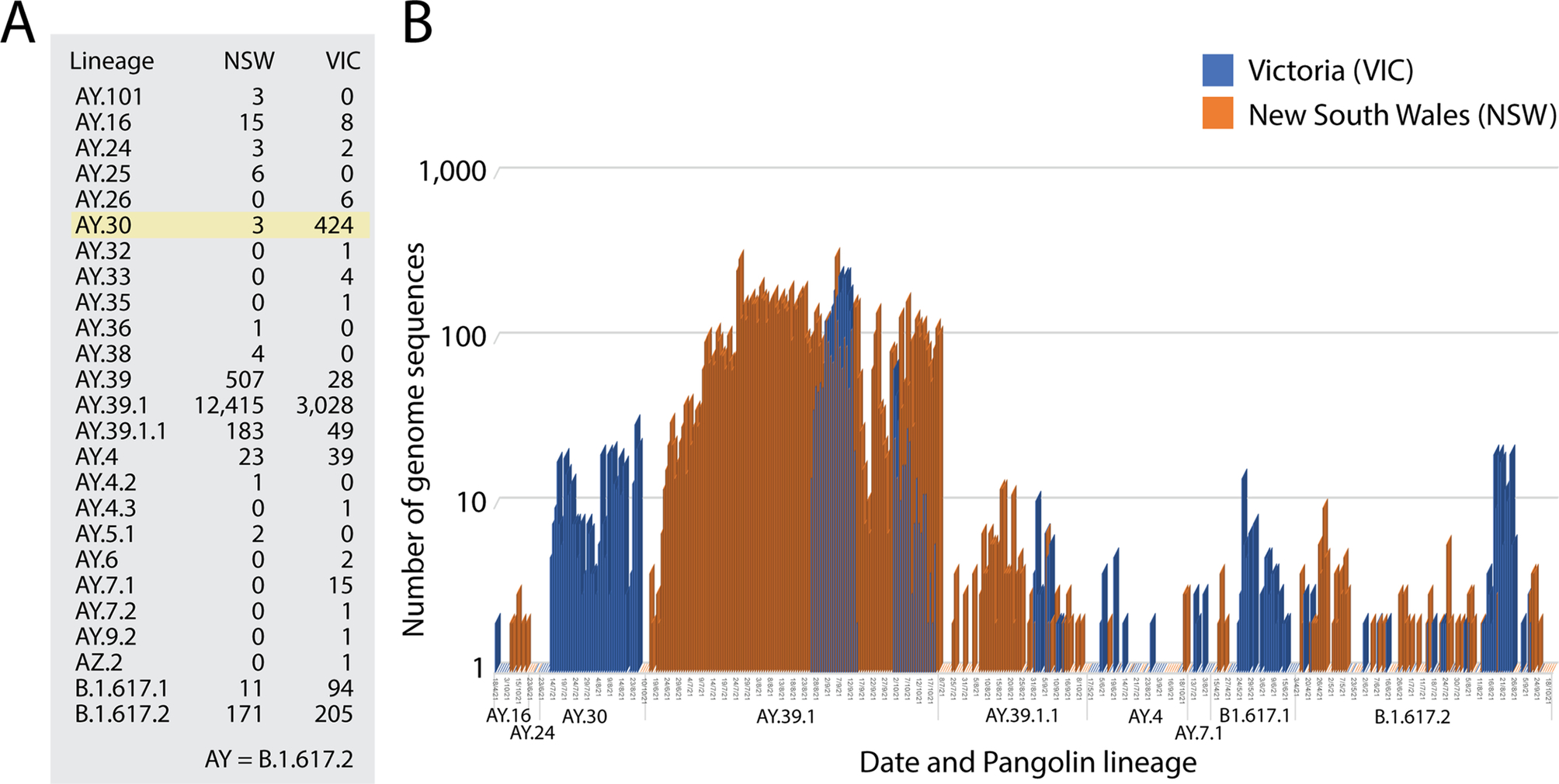 Fig. 2 A seasonal effect during the VOC delta wave of Australia. (A). Assignment of pangolin lineages to 4184 and 13,536 genome sequences from New South Wales (NSW) and Victoria (VIC), respectively. VIC and NSW account for most of VOC delta cases in Australia. (B) The bar plots describe the incidence of pangolin lineages according to date genome sequence acquisition. Lineages assigned to only one sequence are not graphed.
Fig. 2 A seasonal effect during the VOC delta wave of Australia. (A). Assignment of pangolin lineages to 4184 and 13,536 genome sequences from New South Wales (NSW) and Victoria (VIC), respectively. VIC and NSW account for most of VOC delta cases in Australia. (B) The bar plots describe the incidence of pangolin lineages according to date genome sequence acquisition. Lineages assigned to only one sequence are not graphed.
4: Prevalence of amino acid variants in Australia
In order to track the prevalence of individual mutations as they emerged during the entire span of the pandemic, we analyzed 50,744 genome sequences drawn from the 7 regions (state/territories) of Australia, partitioning them into those acquired in each of the 9 calendar quarters. Sequences collected between January 1, 2020 and January 13, 2022 were downloaded on January 18, 2022 (Table 1). From these sequences, a total of 9281 amino acid substitutions (variants) were identified and subsequently filtered with a “relevancy” criterium determined by asserting that substitutions must hit a prevalence threshold of 0.1 (10%) for more than two quarters and in more than two regions. Note that the threshold, number of quarters, and number of regions are dynamic. While there are some limitations to this heuristic, the filtering criterion guarantees that we are not missing any significant mutations, especially those that become VOC “markers.” The figures included in the Supplementary information section show accumulation plots of individual amino acid variants corresponding to the major VOCs appearing in Australia, i.e., VOC alpha (Fig. S1), VOC delta (Fig. S2) and VOC omicron (Fig. S3). Fig. S4 describes accumulation plots of other amino acid variants that were retained following our relevancy criterion. Collectively, the plots describe the set of the most significant mutations that appeared in individual proteins of the viral proteomes in the different regions of Australia and along the 9 quarters of the pandemic. Prevalence ranged from 0 to 1, with 1 implying that 100% of genome sequences collected during an individual quarter contained that mutation. In the following subsection we describe the most salient patterns observed in the accumulation plots.
4.1: The emergence of first haplotypes
Variant accumulation plots showed that the first major haplotypes reported worldwide were also present in Australia (Fig. 3). We found that the D614G amino acid substitution of the spike (S-protein) and the P323L substitution of the NSP12 polymerase that mediates viral replication were coupled (sometimes loosely) in all Australian regions (Fig. 3A). These substitutions are part of a 4-mutation haplotype (labeled here as haplotype 5) that was first established in Europe after its first report in Germany and spread throughout continents during the January–April period of 2020. The haplotype is the most stable so far and is believed to be linked to increases of COVID-19 infectivity (Becerra-Flores & Cardozo, 2020; Korber et al., 2020). Mutations were already present in most Australian regions at 60% prevalence levels during the first quarter of 2020, but at lower levels in Tasmania, ACT and Southern Australia. Remarkably, while these markers of haplotype 5 were maximally prevalent worldwide, their prevalence only reached 100% in the third quarter of 2021 for all regions of Australia. We also note that the emergence of the haplotype was noisy and sometimes decoupled across regions until the second quarter of 2021. Temporal decoupling was evident in the Northern Territory, Queensland, New South Wales, and Western Australia, i.e., regions above the − 34°S latitude transect that are warmer. The slow establishment of haplotype 5 may be explained by the zero-COVID public health policy of the country.
 Fig. 3 The noisy rise of first SARS-CoV-2 haplotypes in Australia. (A). Accumulation plots describing the prevalence of the D614G amino acid substitution of the spike (S) protein and the P323L substitution of the NSP12 polymerase show their joint but noisy emergence and partial decoupling until the second quarter of 2021. Overlapping plots of the two markers make evident the coupling-decoupling patterns in the haplotype (haplotype 5). (B). Accumulation of the R203K and G204R markers of the nucleocapsid (N) protein reveal a tight coupling of the haplotype (haplotype 2) during the start of the pandemic but a later decoupling that began in the last quarter of 2020.
Fig. 3 The noisy rise of first SARS-CoV-2 haplotypes in Australia. (A). Accumulation plots describing the prevalence of the D614G amino acid substitution of the spike (S) protein and the P323L substitution of the NSP12 polymerase show their joint but noisy emergence and partial decoupling until the second quarter of 2021. Overlapping plots of the two markers make evident the coupling-decoupling patterns in the haplotype (haplotype 5). (B). Accumulation of the R203K and G204R markers of the nucleocapsid (N) protein reveal a tight coupling of the haplotype (haplotype 2) during the start of the pandemic but a later decoupling that began in the last quarter of 2020.
The R203K and G204R markers of the nucleocapsid protein (N-protein) emerged as a tightly linked haplotype (haplotype 2) in all regions during the start of the pandemic (Fig. 3B). Mutation decoupling occurred between the third quarters of 2020 and 2021 in Queensland and Victoria. Prevalence reached highest levels between the third quarter of 2020 and the first quarter of 2021 in regions above the − 34°S latitude transect that are colder, especially in ACT, Victoria and Tasmania (which reached 100% prevalence levels), but was found to significantly decrease in regions closer to the Equator. These patterns and the rise of the haplotype during the winter in the Southern Hemisphere suggest a seasonal effect. We note that the haplotype disappeared from Australia in the third quarter of 2021 when VOC delta took over the viral population but later re-emerged forcefully as a tightly linked haplotype with the rise of VOC omicron at the end of 2021. These mutations are located in the serine/arginine-rich linker that separates the N-terminal and C-terminal RNA-binding domains of the N-protein, which we found is intrinsically disordered (Tomaszewski et al., 2020).
4.2: Emergence of haplotypes associated with VOCs alpha, delta and omicron
A classification of accumulation plots revealed the existence of 18 additional haplotypes, which were associated with VOCs alpha, delta and omicron. These haplotypes were composed of 2–12 variants in 1–6 proteins.
The core mutant constellation of VOC alpha was defined by a central haplotype of 12 amino acid variants (haplotype 1), which affected the S-protein, N-protein, the accessory ORF8 immune evasion protein, and the NSP3 papain-like proteinase scaffold. Fig. 4A shows accumulation plots and an overlap plot describing the tight coupling of this large haplotype in Australia between the fourth quarter of 2020 and the third quarter of 2021. The only significant difference in variant accumulation was observed in R52I of ORF8, which started to accumulate in the third quarter of 2020 in South Australia (blue curve in plot overlap). The early appearance of this marker suggests an episode of early recruitment. As expected, prevalence of the haplotype across regions was low (below ~ 60%) since it follows the low prevalence of VOC alpha (see Fig. 1B). However, the haplotype was surprisingly absent in Tasmania.
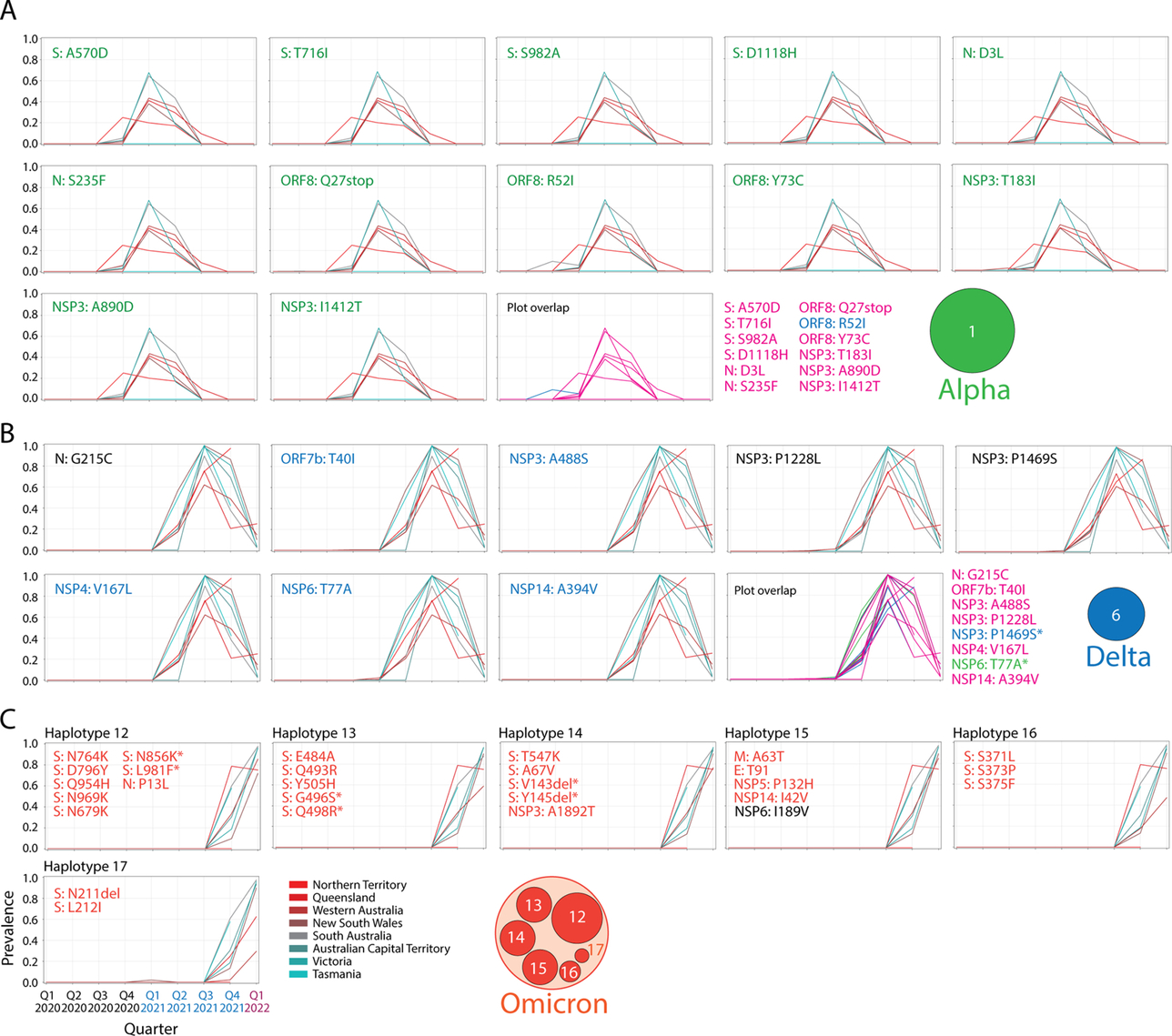 Fig. 4 Core haplotypes of VOC alpha, delta and omicron. (A). Accumulation plots describing the prevalence of the 12 amino acid variants of haplotype 1 of VOC alpha. The plot overlap describes the tight curve overlap of the 12 markers, revealing only a decoupling pattern in the curves of variant R52I of the ORF8 protein. (B). Accumulation plots describing the prevalence of the 8 amino acid variants of haplotype 6 of VOC delta. The “plot overlap” describes the tight curve overlap of the 7 markers. Note that there was decoupling associated with variants P1469S of NSP3 and T77A of NSP6. (C) Accumulation plots describing the prevalence of representative variants (first in the lists) of the 6 haplotypes that make up the core of VOC omicron (haplotypes 12–17). For all panels, plots describe variant accumulation in the 8 regions of Australia and are labeled with the name of the amino acid variant colored according to its presence in the mutant constellation reported for VOCs worldwide (green, VOC alpha; blue, VOC delta; red, VOC omicron; black, other). The icons for the alpha, delta and omicron haplotype cores are being used in the network of Fig. 5.
Fig. 4 Core haplotypes of VOC alpha, delta and omicron. (A). Accumulation plots describing the prevalence of the 12 amino acid variants of haplotype 1 of VOC alpha. The plot overlap describes the tight curve overlap of the 12 markers, revealing only a decoupling pattern in the curves of variant R52I of the ORF8 protein. (B). Accumulation plots describing the prevalence of the 8 amino acid variants of haplotype 6 of VOC delta. The “plot overlap” describes the tight curve overlap of the 7 markers. Note that there was decoupling associated with variants P1469S of NSP3 and T77A of NSP6. (C) Accumulation plots describing the prevalence of representative variants (first in the lists) of the 6 haplotypes that make up the core of VOC omicron (haplotypes 12–17). For all panels, plots describe variant accumulation in the 8 regions of Australia and are labeled with the name of the amino acid variant colored according to its presence in the mutant constellation reported for VOCs worldwide (green, VOC alpha; blue, VOC delta; red, VOC omicron; black, other). The icons for the alpha, delta and omicron haplotype cores are being used in the network of Fig. 5.
The core mutant constellation of VOC delta involved an 8-variant haplotype (haplotype 6) affecting 6 proteins—the N-protein, ORF7b, the NSP3 protease, NSP4, NSP6 and the NSP14 exonuclease (Fig. 4B). Minor differences in variant accumulation were observed in P1469S of NSP3 (most notably in Victoria and the Northern Territory) and in T77A of NSP6 (in South Australia and the Northern Territory). Remarkably, haplotype prevalence reached 90–100% in regions below the − 34°S latitude transect (South Australia, ACT, Victoria and Tasmania) during the third quarter of 2021, while for example, it reached only 60% in New South Wales.
Finally, the core mutant constellation of VOC omicron involved a set of 6 haplotypes (haplotypes 12–17) containing 2–8 variants, each of which affected 1–5 proteins, including the S-protein, N-protein, and the membrane (M) and envelope (E) structural proteins, NSP3, NSP5, NSP6 and NSP14 (Fig. 4C). Haplotypes 13, 16 and 17 affected sites exclusively present in the S-protein. Haplotypes 12 and 14 also involved a significant number of S-protein markers. Accumulation curves showcase how VOC omicron overtook the entire viral population in all regions and in a period of only two calendar quarters, the last quarter of 2021 and the first quarter of 2022. Curve overlaps for variants in each haplotype revealed an absence of significant decoupling patterns. We note, however, that all six haplotypes harbored common patterns of accumulation, which suggests they exhibit similar behavior across regions. This merits placing these haplotypes within a single haplotype core. We also note that S-protein variants A67V and V143del of haplotype 14 and N210del and N212I of haplotype 17 appeared in advance of VOC delta in the first quarter of 2021, which suggests that these markers were recruited from markers that were already present in the viral population in early 2021. Furthermore, the N-protein variant P13L of haplotype 12, which is associated with the N-terminal region of the nucleocapsid that is intrinsically disordered (Tomaszewski et al., 2020), appeared during the first wave of the pandemic in the first quarter of 2020, reaching very high prevalence levels in Tasmania. This marker, which was part of a predicted pathway of mutational change involving protein flexibility/rigidity (Tomaszewski et al., 2020), likely represents the oldest variant of the VOC omicron haplotype core.
4.3: Amino acid variants and haplotypes shared by VOCs
Out of all variants and haplotypes identified in our analysis of mutational prevalence, only a handful were shared between VOCs in Australia. Out of a total of 98 markers, 74 were present in haplotypes but only 7 of these were shared between two or three VOCs. They defined the only 4 shared haplotypes (out of a total of 20) described above. Conversely, 6 out of 24 single-standing variants were shared between two VOCs. These numbers suggest limited but yet significant recruitment during VOC emergence in episodes of variant and haplotype coalescence.
Haplotypes 2, 3 and 4, as well as single-standing variants P681H and N501Y of the S-protein were shared between VOCs alpha and omicron. Haplotype 2 (described above) involved two mutations in the central intrinsically disordered linker of the N-protein, haplotype 3 involved three deletions in the N-terminal domain (NTD) of the S-protein (H69del, V70del, and Y144del), and haplotype 4 involved two deletions (S106del and G107del) in NSP6. In turn, only 4 variants unified VOCs delta and omicron, i.e., variants S96I, G142D and T478K of the S-protein and variant T492I of NSP4. Only the most ancestral haplotype, haplotype 5 mentioned above, unified all three VOCs.
4.4: Amino acid variants that are not part of established VOC constellations
We identified a number of amino acid variants with significant prevalence that did not belong to established VOC constellations (see CoVariants; https://covariants.org). Accumulation plots are shown in Fig. S4 in Supplementary information. Mutants E484K and A701V of the S-protein, T205I of the N-protein, T85I of NSP2, K835N of NSP3, and K90R of NSP5 followed increases that mirrored those of VOC alpha markers. Similarly, G215C of the N-protein and V71I of ORF7a followed increases similar to those of VOC delta. Finally, V1069 of NSP3 increased together with the emergence of VOC omicron. Mutants with increases that mirrored those of VOC alpha markers reached 100% prevalence in ACT during the last quarter of 2020. Another set reached prevalence in ACT during the second quarter of 2021 (R385K of the N-protein, P129L of NSP2, H1274Y of NSP3, and H234Y of NSP15). We cannot explain why the ACT region fostered all of these mutations at high levels. The high prevalence reached by S197L of the N-protein and F308Y of NSP4 in Tasmania cannot be explained either, especially because both mutations had patterns of accumulation that were linked (suggesting an haplotype). They may represent specific mutational bursts that occurred in those regions.
5: A network view of haplotype diversity and VOC emergence
A network view can help better describe the haplotype and variant makeup of VOCs. Fig. 5 shows a “haplotype network” describing the viral population landscape of Australia that unfolded throughout the COVID-19 pandemic. Nodes of the graph are either haplotypes or individual amino acid variants coalescing into VOC-specific constellations. Edges describe common patterns of prevalence in accumulation plots. Circles portray levels of haplotype coalescence. Outer-most circles host amino acid variants and haplotypes shared between VOCs. Circles closer to the core haplotypes host variants and haplotypes with patterns of prevalence that resemble more tightly those of the core. The network reveals significant and unanticipated patterns of emergence and diversification, which we discuss in the following subsections.
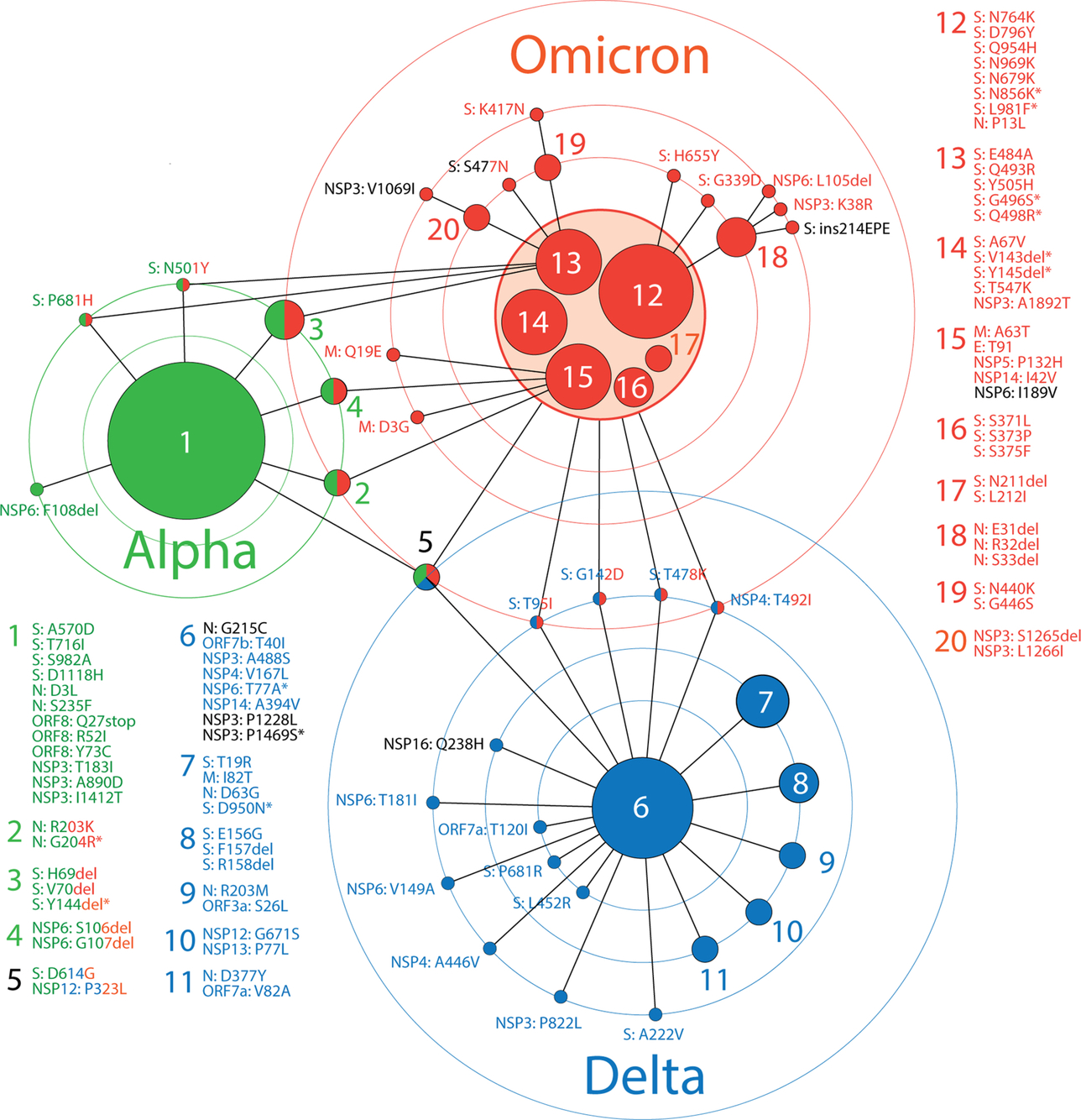 Fig. 5 A network of haplotypes describes the emergence of major VOCs in Australia. Nodes are either haplotypes or individual amino acid variants coalescing into VOC-specific constellations. Edges describe common patterns of prevalence in accumulation plots. Circles portray levels of haplotype coalescence. Outer-most circles host amino acid variants and haplotypes shared between VOCs. Haplotypes are labeled with numbers and variants are labeled with names that follow accepted nomenclature from the Human Genome Variation Society. Names are colored according to their presence in established VOCs worldwide or in black when uniquely present in Australia.
Fig. 5 A network of haplotypes describes the emergence of major VOCs in Australia. Nodes are either haplotypes or individual amino acid variants coalescing into VOC-specific constellations. Edges describe common patterns of prevalence in accumulation plots. Circles portray levels of haplotype coalescence. Outer-most circles host amino acid variants and haplotypes shared between VOCs. Haplotypes are labeled with numbers and variants are labeled with names that follow accepted nomenclature from the Human Genome Variation Society. Names are colored according to their presence in established VOCs worldwide or in black when uniquely present in Australia.
5.1: Haplotype and variant reuse
The currently widespread VOC omicron appears to have drawn markers from haplotypes and variants of VOC alpha more than from VOC delta. With the exception of the ancestral haplotype 5 typical of all three VOCs, VOC omicron shares 3 haplotypes with VOC alpha (haplotypes 2, 3 and 4) involving markers of the S-protein, N-protein and NSP6, respectively. In turn, VOC omicron shares only 4 variants with VOC delta. These markers, 3 of which are S-protein variants, coalesce into VOC delta's core. In other words, VOC alpha contributed almost half of its markers (11 out of 24 total) and 4 out of its 5 haplotypes to VOC omicron, while VOC delta contributed only 17% of its markers (6 out of 35 total) and only 1 out of its 7 haplotypes to the makeup of the new VOC.
Going back in time, a number of omicron-specific variants were already present in significant number during the first wave of the pandemic early in 2020. These include the S477N variant of the S-protein and the P13L variant of the N-protein. Other omicron-specific markers appeared by the end of 2020 and beginning 2021 in Australia, including K417N and to a lesser level A67V, N440K, H655Y, N679K, D796Y of the S-protein and K38R, S1265del, and L1266I of NSP3 (Fig. S3 in Supplementary information). Similarly, several delta-specific variants were already present in 2020 and very early in 2021, including A222V, L452R and P681R of the S-protein, 182 T of the M-protein, D377Y of the N-protein, P822L of NSP3, A446V of NSP6, T181I of NSP6 and to a lesser level V82A of ORF7a, T40I of ORF7b, P822L of NSP3, P1228L of NSP3, and P77L of NSP13 (Fig. S2 in Supplementary information). Finally, alpha-specific variants were already significantly present during the end of the first wave, including R52I of ORF8 and T183I of NSP3 (Fig. S1 in Supplementary information). Many of these reused markers were part of several haplotypes.
In order to strengthen the argument of significant reuse of markers in variant combinations, we explored their presence in 137,605 sequences of the S-protein retrieved worldwide on November 14, 2020 (Showers, Leach, Kechris, & Strong, 2022). For VOC omicron, D614G of haplotype 5, H69del, V70del and Y144del of haplotype 3 (shared with VOC alpha), D796Y and N679K of haplotype 12, A67V, Y143del and T547K of haplotype 14, and N440K of haplotype 19, were present in the dataset sometimes in combination with others. For example, H69del and V70del variants of haplotype 3 were present in 1066 genomes as a H69del-V70del-N439K-D614G combination and in numerous other arrangements in another 698 sequences, including 22 sequences of a 10-variant combination of 6 VOC omicron-specific and 3 VOC-alpha-specific markers (H69del-V70del-Y145del-N501Y-A570D-D614G-P681H-T716I-S982A-D1118H). Thus, 5 out of 8 haplotypes affecting S-proteins in VOC omicron recruited markers appearing in 2020. Other free-standing markers were also recruited, including S477N, D574Y, G339D, G142D and T478K (both shared with VOC delta), and P681R (shared with VOC alpha). In particular, S477N was present in 8080 sequences as the second most popular combination of the set (S477N-D614G). For VOC delta, T19R and D950N of haplotype 7 (the only S-protein markers in haplotypes), G142D and T478K shared with VOC omicron, P681R, L452R and A222V were also present in the dataset. In particular, A222V appeared in 7088 sequences as a L18F-A222V-D614G combination and in 169 sequences as a L5F-A222V-D574Y-D614G-H655Y combination (which includes H655Y of VOC omicron). As expected for VOC alpha, we found 22 instances of the 10-variant combination containing all S-protein markers of haplotype 1 (A570D, T716I, S982A, D1118H), haplotype 3 (H69del, V70del, Y145del) and haplotype 5 (D614G) and the two free-standing variants N501Y and P681H that collectively characterize the S-protein constellation of this viral variant (H69del-V70del-Y145del-N501Y-A570D-D614G-P681H-T716I-S982A-D1118H). Recall that the first appearance of VOC alpha occurred a few weeks before the sampling date of the dataset in the United Kingdom. To summarize, 16 S-protein variants of VOC omicron, 7 of VOC delta and all variants of VOC alpha were already present before November 2020. The appearance of 10-variant combinations of markers of VOCs delta and alpha is particularly significant and suggests the existence of massive viral recruitment, perhaps mediated by recombination.
5.2: Haplotype size and coalescence
Results of our analyses reveal an apparent correlation between haplotype size, VOC age and coalescence. VOC alpha emerged earlier than VOCs delta and omicron in both Australia and the rest of the world (Fig. 1B). Half of the 24 variants of VOC alpha coalesced into the largest known haplotype, haplotype 1. With the exception of 3 variants, the rest coalesced into 4 additional haplotypes, 3 of which had accumulation patterns that resembled those of the core. Conversely, 23 out of the 36 variants of VOC delta coalesced into 7 haplotypes, the largest of which (haplotype 6) had a substantially smaller number of markers than haplotype 1 of VOC alpha. A total of 13 variants remained unlinked, suggesting a lower level of haplotype coalescence operating during the reign of VOC delta. VOC omicron originated very recently. It involved recruitments of many VOC-specific markers that appeared quite early in the pandemic (especially the N-protein variant P13L of haplotype 12). As expected, the levels of coalescence of VOC omicron are the lowest of the three VOCs judged by a non-unified constellation core of 6 haplotypes, the existence of 7 peripheral haplotypes, and 14 additional unlinked markers. In particular, the 28 markers of the constellation core, which harbor quite distinct accumulation patterns (Fig. 4), represent only about half of the 58 markers of VOC omicron in Australia. Thus, older haplotypes are larger and exhibit higher levels of coalescence, assuming they have not been completely replaced by incoming haplotypes of VOCs and are part of a growing pool of viral genetic diversity.
5.3: Haplotypes and protein interactions
We have observed distinct groups of proteins that have been mutated in the different VOCs. The core haplotypes of VOCs alpha and delta involved a diverse set of proteins, while those of VOC omicron are now highly enriched in mutations affecting the S-protein. Fig. 6 shows a network of SARS-CoV-2 proteins with links describing their joint presence in haplotypes. Pie charts representing selected nodes of the network describe how intramolecular interactions define haplotypes within individual molecules. Since haplotypes typically arise by evolutionary constraints imposed on protein-protein interactions, intramolecular interactions (e.g., allosteric interactions), or indirectly through shared or linked functions, the network suggests how the creation of mutant constellations contribute to the gradual enhancement of molecular interactions that benefit viral persistence. In the network, VOC alpha interactions (lines colored in green in the graph) involving the spike and nucleocapsid structural proteins were extended by interactions involving a number of nonstructural and accessory proteins in VOCs delta and omicron (lines in blue and red, respectively). The central S-protein, N-protein and NSP3 protease connection established via multiple markers of haplotype 1 in VOC alpha was atomized in the haplotypes of VOC delta but was later (and forcefully) regained in VOC omicron through S-protein links to both N-protein (haplotype 12) and NSP3 (haplotype 14), N-protein-specific haplotype 18, NSP3-specific haplotype 20, and 6 core haplotypes with a multiplicity of S-protein markers. This solidification of the functionalities of the spike, nucleocapsid and NSP3 papain protease in VOC omicron makes evident their well-known centrality in viral transmissibility, disease severity, and immune escape. The S-protein plays critical roles in viral attachment to host cells. Its highly immunogenic properties and roles in transmissibility and virulence has made the spike glycoprotein trimer a target for drug and vaccine mitigation (Harvey et al., 2021). The N-protein, which packages the RNA genomes, is the most abundant viral protein and is essential for replication, virion assembly, and regulation of the viral life cycle (Bai, Cao, Liu, & Li, 2021). In addition, the two structural domains of the N-protein are separated by an intrinsically disordered linker that is highly sensitive to proteolysis and generates at least five proteoforms that bind structured RNA (Lutomski, El-Baba, Bolla, & Robinson, 2021). This endows the N-protein with a host of regulatory and immunogenic properties. Finally, the multidomain NSP3 papain-like protease acts on the viral polyproteins, interacts with other NSPs and RNA to form the replication/transcription complex, antagonizes the host innate immune response, and supports viral survival (Lei, Kusov, & Hilgenfeld, 2018). The central role of these three proteins is enhanced in VOCs by an additional central hub associated with authophagy, the NSP6 protein (Fig. 6). NSP6 has been shown to induce autophagosome formation and NLRP3 inflammasomes, mediating caspase-1 activation and secretion of pro-inflammatory cytokines known to induce inflammatory cell death (Cottam et al., 2011; Sun, Huang, Xu, & Hu, 2021). The inflammasomes are multimeric sensor proteins that are critical components of the innate immune system. However, their aberrant activation can cause serious disorders, including cascades leading to the severe acute respiratory syndrome (SARS) caused for example by SARS-CoV-2 (Rodrigues et al., 2021). The NSP6-specific haplotype 4 of VOC alpha shows an early central role in this aberrant activation of the innate immune system, which was later complemented by the delta-specific core haplotype 6 and haplotype 9, which are linked to N-protein and ORF3a. Remarkably, the ORF3a viroporin has been shown to inhibit autophagosome-lysosome fusion by interacting with a protein of the homotypic fusion and protein sorting (HOPS) complex (Zhang et al., 2021). This mechanism helps the virus escape degradation. The central role of NSP6 continues in VOC omicron with markers of haplotype 4, haplotype 15 and an extra free-standing NSP6 marker (Figs. 5 and 6), which prompts evaluation of how VOC omicron mutations are softening aberrant immunity activations.
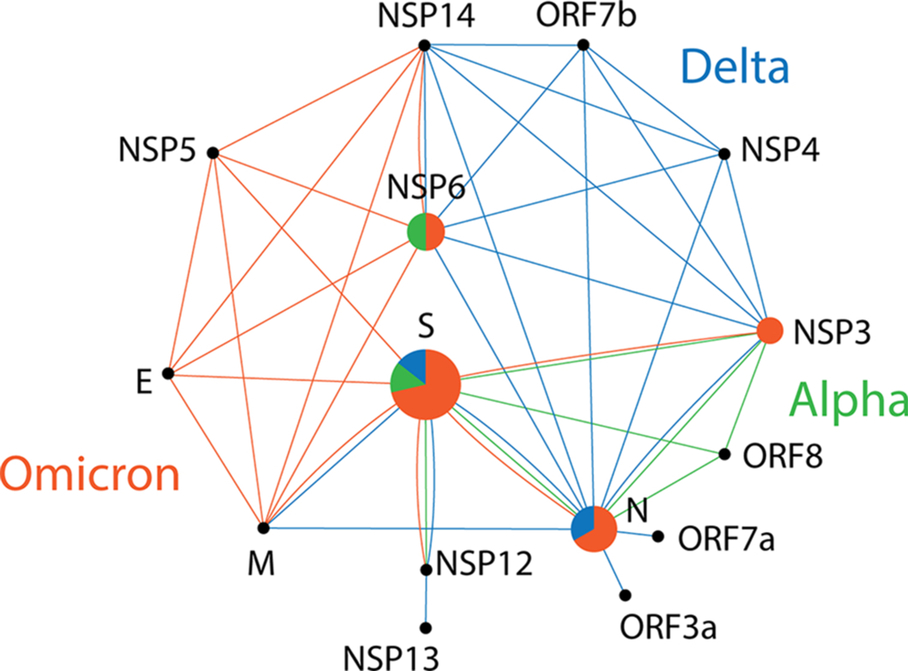 Fig. 6 A network of SARS-CoV-2 proteins mediated by haplotypes. Nodes are proteins and lines of the graph are protein interaction expressed as joint protein presence in an haplotype. Pie charts are nodes describing the relative number of haplotypes made up of only variants of that protein in the mutant VOC constellations. Pie slices and lines of the graph corresponding to VOC alpha, delta and omicron are colored green, blue and red, respectively.
Fig. 6 A network of SARS-CoV-2 proteins mediated by haplotypes. Nodes are proteins and lines of the graph are protein interaction expressed as joint protein presence in an haplotype. Pie charts are nodes describing the relative number of haplotypes made up of only variants of that protein in the mutant VOC constellations. Pie slices and lines of the graph corresponding to VOC alpha, delta and omicron are colored green, blue and red, respectively.
The rise of VOC delta haplotypes appear to optimize interactions of the N-protein with the M-protein and S-protein, and VOC omicron haplotypes similarly now optimize interactions between the M-protein and both the E-protein and the S-protein (Fig. 6). Intraviral interactions between these three structural proteins have been reported to play essential roles in the viral life cycle (e.g., Artika, Dewantari, & Wiyatno, 2020). They hijack the cellular network of the host. The central intrinsically disordered serine/arginine-rich spacer of the N-protein that separates its two domains is vital for effective viral replication. The transmembrane M-protein consist of an N-terminal ectodomain and a C-terminal endodomain. The endodomain interacts with multiple regions of both the N-protein and S-protein for oligomerization, RNA encapsulation and mature virus particle formation but also with the E-protein through two transmembrane and the cytoplasmic domains (Hsieh, Li, Chen, & Lo, 2008). The interactions of the M-protein with the S-protein, E-protein and N-protein of SARS-CoV-2 have been recently modeled using protein-protein docking and molecular dynamic simulation (Kumar, Kumar, Garg, & Giri, 2021). The M protein acts as receptor, while the E-protein, N-protein and S-protein act as protein ligands. For example, transient helices in the domains and linkers of the N-protein establish interactions with a number of proteins, including the M-protein (Lu et al., 2021) and NSP3 of the viral-replicase transcription complex (Hurst, Koetzner, & Masters, 2013). Clearly, the haplotype-mediated network of proteins shown in Fig. 6 makes these interactions evident in VOC evolution.
5.4: VOC omicron haplotype variants cluster along the S-protein sequence
Given the centrality of the spike in viral-host recognition and the enrichment of S-protein variants in the mutant constellation of VOC omicron, we explored the location of omicron-specific mutations along the sequence of the S-protein. We asked if amino acid substitutions in haplotypes were clustered in domains along the sequence. Remarkably, Fig. 7 shows haplotypes markers grouped around defined regions of the S-protein. Most variants in haplotypes 3, 14 and 17 mapped to the N-terminal domain (NTD) while those of haplotypes 13, 16 and 19 did so to the receptor binding domain (RBD). In particular mutations in the receptor-binding motif (RBM) correspond to those of haplotype 13. Mutations affecting the S2 transmembrane subunit were part of haplotype 12. The 8 free-standing mutations (labeled in black) mapped to other positions but were tightly associated with haplotypes. For example, RBM-linked N501Y and S477N and markers of haplotype 13 had common patterns in accumulation plots and were close in protein location (Fig. 5). Similarly, H655Y and haplotype 12 were also similarly associated and so did K417N and haplotype 19. Thus, markers coalesce into haplotypes following clustering patterns in defined regions of the S-protein.
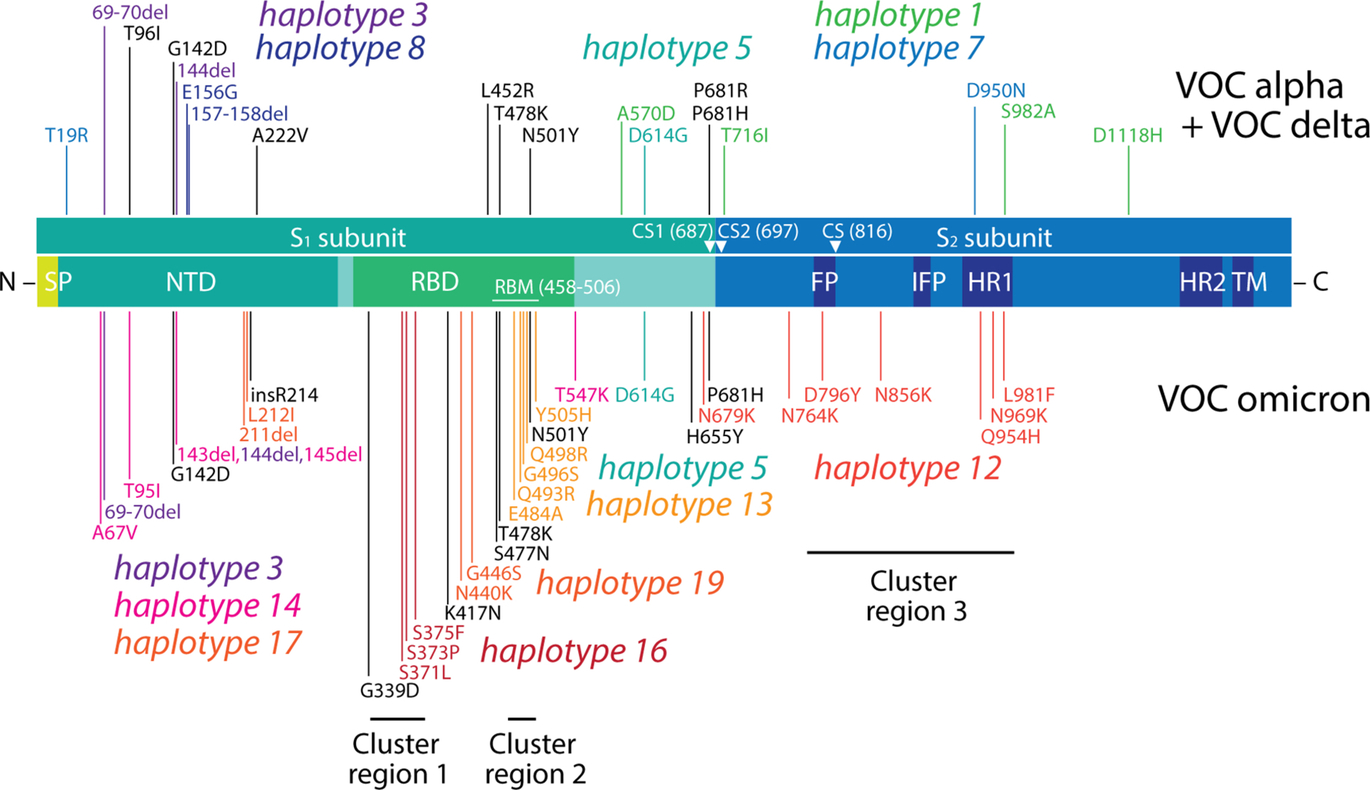 Fig. 7 Mutations of the S-protein characteristic of VOC omicron cluster in groups according to haplotype and are enriched in immune evasion functions associated with the RBD region. The diagram maps mutations onto the different regions of the S-protein molecule from N-terminus to C-terminus in the amino acid sequence, with markers specific to VOCs alpha and delta indicated in the top and those specific for VOC omicron in the bottom. Clusters 1, 2 and 3 represent mutations arising from nucleotide substitutions at codon sites that are either negatively selected or are evolving under no detectable selection in non-omicron sequences. SP, signal peptide; NTD, N-terminal domain; RBD, receptor-binding domain; RBM, receptor-binding motif; CS, cleavage site; FP, fusion peptide; IFP, internal fusion peptide; HR1, heptad repeat 1; HR2, heptad repeat 2; TM, transmembrane domain.
Fig. 7 Mutations of the S-protein characteristic of VOC omicron cluster in groups according to haplotype and are enriched in immune evasion functions associated with the RBD region. The diagram maps mutations onto the different regions of the S-protein molecule from N-terminus to C-terminus in the amino acid sequence, with markers specific to VOCs alpha and delta indicated in the top and those specific for VOC omicron in the bottom. Clusters 1, 2 and 3 represent mutations arising from nucleotide substitutions at codon sites that are either negatively selected or are evolving under no detectable selection in non-omicron sequences. SP, signal peptide; NTD, N-terminal domain; RBD, receptor-binding domain; RBM, receptor-binding motif; CS, cleavage site; FP, fusion peptide; IFP, internal fusion peptide; HR1, heptad repeat 1; HR2, heptad repeat 2; TM, transmembrane domain.
All mutations of VOC omicron are under gene-wide positive selection (Viana et al., 2022). In a recent study, however, mutations in the S-protein amenable to natural selection analysis methods that focus on patterns of synonymous and non-synonymous mutations (together with patterns of mutational convergence and prevalence) revealed 13 mutations arising from nucleotide substitutions at codon sites that were either negatively selected or were evolving under no detectable selection in non-omicron sequences (Martin et al., 2022). Mutations at these sites were likely interacting with each other, were collectively adaptive, and may be imposing functions typical of the VOC omicron viral population. Remarkably, these mutations clustered into three distinct groups of epistatically-interacting substitutions, which we highlight in Fig. 7. These clusters clearly correspond to haplotypes defined in our study. Cluster 1 mutations, which affect the N-terminal region of RBD, are part of haplotype 16. Cluster 2 mutations, which affect the RBM region of RBD, are part of haplotype 13. Finally, cluster 3 mutations, which affect the fusion domain of the S2 subunit, are part of haplotype 12. Such congruence strengthens our conclusions.
5.5: Haplotypes follow the three phases of the COVID-19 pandemic
The timeline of clades and VOCs shows three successive phases driven by proteome flexibility/rigidity, environmental sensing, and vaccine-driven immune escape (Fig. 1). These proposed phases were based mostly on mutation bursts appearing in the S-protein (Caetano-Anollés et al., 2022). We find that the development of haplotypes with S-protein markers (Fig. 7) faithfully followed the phases of the timeline (Fig. 1).
6: Continental links to seasonality
Effective COVID-19 transmission appears restricted to a 30°N to 50°N latitude corridor in both the Northern and Southern Hemispheres (Caetano-Anollés et al., 2022). Here we explore differences in mutation accumulation patterns of haplotypes and free-standing markers that exist between regions of Australia that span different latitudes, revealing a continental link to viral seasonal behavior. In our analysis, calendar quarters are able to coarse-grain general patterns of prevalence. This feature allows to visualize region-specific patterns. Because the Australian continent spans a roughly − 10°N to − 45°N latitude corridor, we used a –34°S latitude transect to dissect regions 1–4 that are closer to the Equator (from the Northern Territory to New South Wales) from the colder and more southern regions 5–8 (from South Australia to Tasmania). This transect separates the largest cities of Sydney and Melbourne from each other. These cities have been major contributors to cases in Australia, with Melbourne contributing to more deadlier outcomes.
6.1: Core haplotypes of VOCs reveal latitude-linked patterns of seasonality
We first concentrated on core haplotypes of VOCs alpha, delta and omicron (Fig. 8). We excluded haplotypes arising before the first appearance of VOCs, i.e., the D614G containing haplotype 5, which was recruited by all three VOCs, and the N-protein-specific haplotype 2, which was recruited by VOCs alpha and omicron. In the case of VOC delta we retained all six VOC-specific haplotypes and in the case of VOC omicron we retained only the central 6-haplotype core. We then analyzed mutant accumulation curves of all markers belonging to these haplotypes separately for regions 1–4 and regions 4–8. To make differences explicit, we overlapped curves of the four regions in each subset. We reasoned that if markers were tightly linked to each other in an haplotype, curve overlaps would show minimum differences in accumulation between regions. Alternatively, if markers were decoupled then curves would show idiosyncratic patterns. Remarkably, we found significant decoupling patterns in the curves of regions 1–4, which lie outside of the seasonality-linked − 30°N to − 50°N latitude corridor.
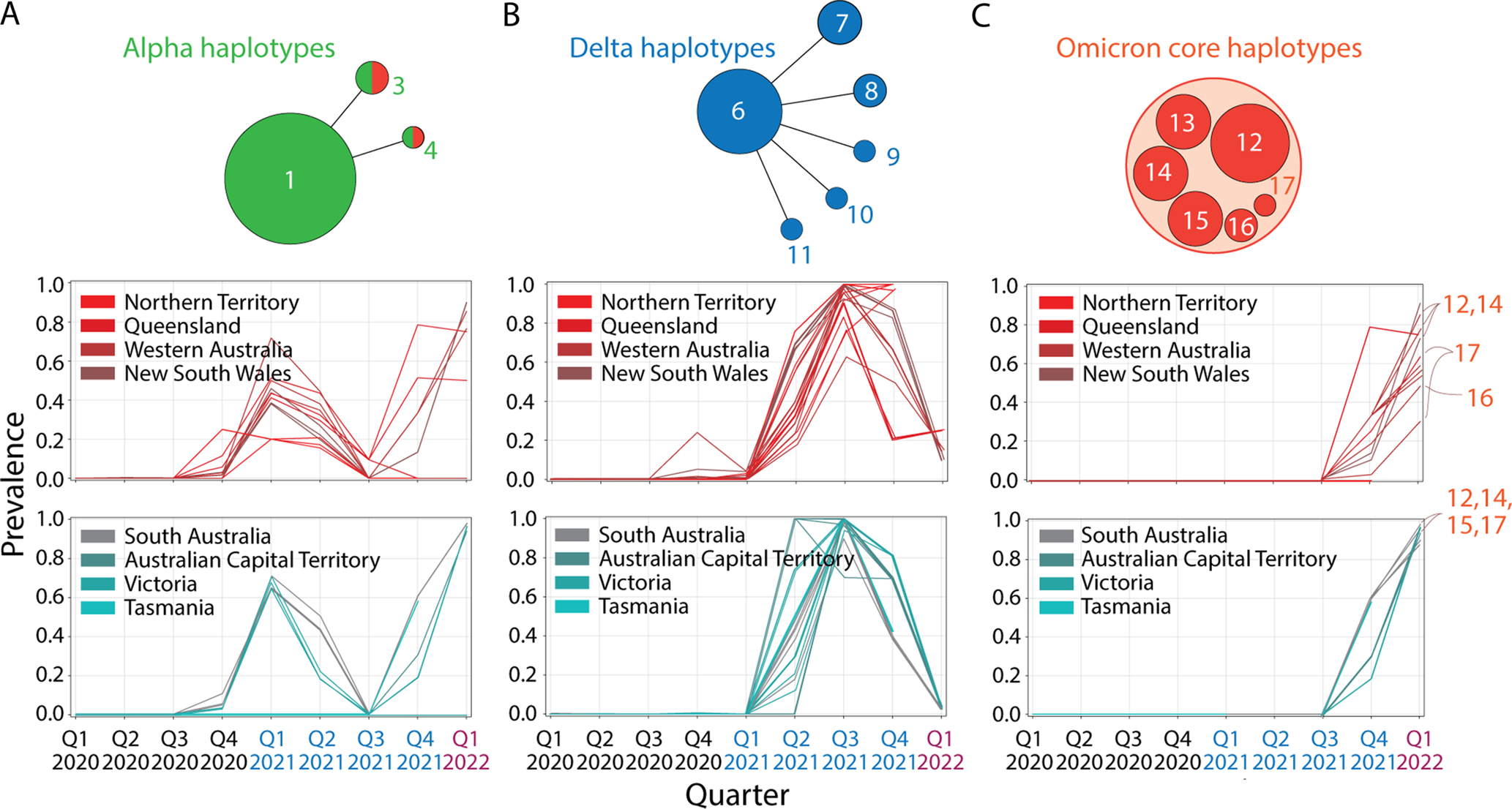 Fig. 8 Patterns of mutation accumulation in core VOC haplotypes reveal seasonal behaviour.
Fig. 8 Patterns of mutation accumulation in core VOC haplotypes reveal seasonal behaviour.
In the case of VOC alpha (Fig. 8A), the incidence of markers in regions 1–4 was highly variable, as showcased by the multiplicity of accumulation patterns (curves). As an example, regions 1–4 displayed 12 distinct curves in quarters 1 and 2 of 2021, while regions 5–8 displayed only 5. Haplotypes shared with VOC omicron (haplotypes 3 and 4) unfolded between the third quarter of 2021 and the first quarter of 2022 significant variability in regions 1–4 (7 curves) compared to those of regions 5–8 (4 curves).
When considering haplotypes of VOC delta, the distinction between accumulation patterns became significant (Fig. 8B). A focus on the second quarter of 2021 revealed 16 distinct curves for region 1–4 versus 14 curves for region 5–8. As the prevalence of VOC delta decreased two calendar quarters later, 12 curves were evident for regions 1–4 while 6 existed for regions 4–8. This suggests VOC delta haplotypes show a pattern of increased coalescence in regions spanning the latitude corridor of seasonality. Please note that VOC delta harbored relatively few markers in the S-protein when compared to the other VOCs, suggesting seasonal patterns also arise from multi-protein interactions.
Finally, the rise of VOC omicron again shows a clear distinction between corridor and non-corridor regions (Fig. 7C). Note, however, the significant diversity of accumulation patterns that exists below the − 34°S latitude transect and the highly coupled patterns above that latitude in colder regions of Australia. The distinction is particularly relevant because of the many haplotypes that make up the VOC omicron core.
6.2: Free-standing markers also support seasonal behavior in Australia
The accumulation of the T95I, G142D and T478K free-standing markers of the S-protein and the T492I marker of NSP4, all of which are shared by VOCs delta and omicron, are particularly insightful (Fig. 9). Markers accumulated significantly in regions 5–8 (especially in South Australia, Victoria and Tasmania) in the second and third quarters of 2021 (winter) while significant accumulation in regions 1–4 were only evident in the fourth quarter of 2021, a time that coincided with the rise of VOC omicron. Curiously, T95I started to accumulate earlier than the other markers but at low prevalence levels (~ 10% levels) in regions 1–4. We postulate all these markers are sensor proteins recruited by VOCs delta and omicron. Their accumulation support the genetic differences we detected between New South Wales and Victoria in Fig. 2.
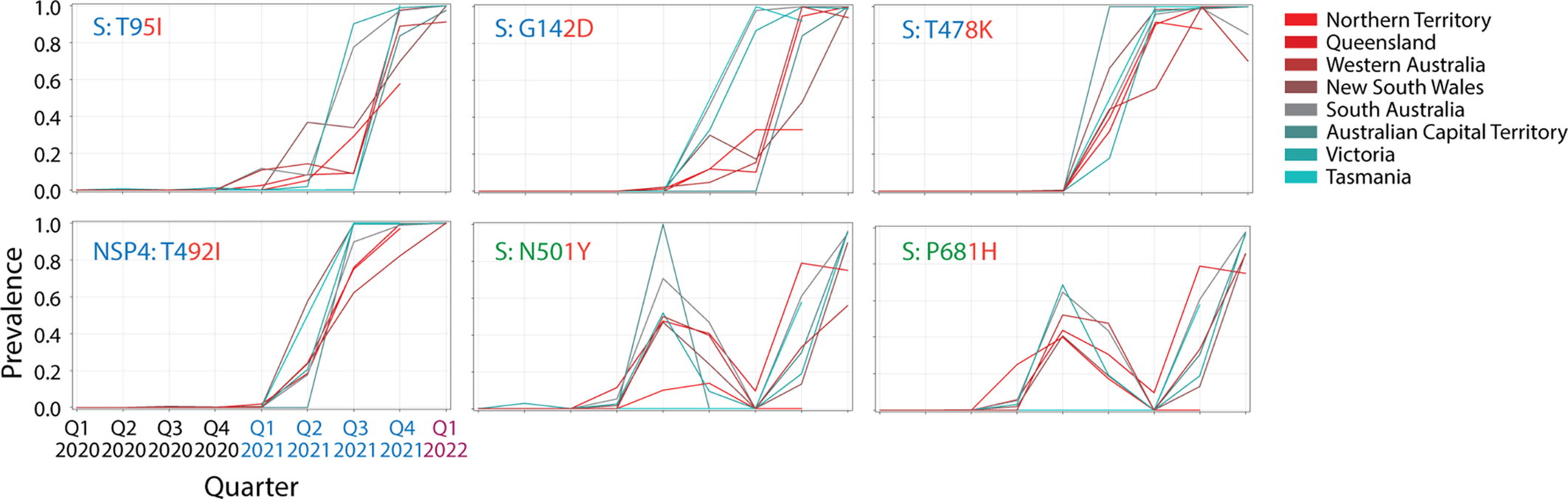 Fig. 9 Free-standing mutations shared by VOCs also show unexpected links to seasonality.
Fig. 9 Free-standing mutations shared by VOCs also show unexpected links to seasonality.
The temporal accumulation of the N501Y and P681H markers of the S-protein that are shared by VOCs alpha and omicron was also interesting. Prevalence peaked during the fourth quarter of 2020 reaching 60–100% levels in South Australia and ACT, disappearing in the third quarter of 2021 and quickly increasing with the rise of VOC omicron. Again, accumulation in regions 1–4 was in general lower, with the exception of Queensland. Out of all 6 markers, T478K was the first to reach and maintain 100% prevalence levels, doing so in the second quarter of 2021 at a time VOC delta was just starting to accumulate in the planet. All of these rather noisy patterns of accumulation suggest VOCs engaged in latitude-dependent recruitment of markers that were already significantly present in the early viral population.
7: Discussion
A recent opinion article claims there is no “transparent” path of transmission linking VOC omicron to its predecessors and no explanation for the unusual array of mutations that appeared to have evolved outside scrutiny of researchers (Mallapaty, 2022). Indeed, VOC omicron was able to quickly develop a large constellation of mutations affecting not only the S-protein but also many other proteins of functional significance (Viana et al., 2022). Similarly, its appearance out of thin air was unanticipated and puzzling. Our results, however, do show that many of VOC omicron mutations were already present during the first year of the pandemic. They were recruited piecemeal a year later to be part of a complex mutant constellation. In fact, 16 S-protein variants of VOC omicron, 7 of VOC delta, and all variants of VOC alpha were already present before November 2020 forming for example combinations of 10 variants in a genome sequence. Some of these mutant combinations were present in large numbers in the viral population that was sampled for sequencing. These results strongly support the existence of massive viral recruitments occurring already during the first waves of the pandemic. The fact that we found that the molecular profile of VOC omicron did not appear monolithically in different regions of Australia also challenges two other theoretical explanations for the origin of VOC omicron (Mallapaty, 2022), namely that all mutations evolved in one patient as part of a long-term infection or that their emergence occurred unseen in other animal hosts (e.g., rodents). These scenarios, which are more compatible with evolutionary founder effects and super-spreader events are therefore unlikely.
VOC omicron was first detected by a genomic surveillance team in November 2021 after a resurgence of infections in the Gauteng Province of South Africa. By mid-December, this new VOC was present in 87 countries in patterns that suggested rapid transmission in regions with high levels of population immunity. Such fulminant appearance was tracked with a time-calibrated Bayesian phylogenetic analysis that placed the origin of the most recent common ancestor to October 9, 2021 and revealed that viral spread from the Gauteng province to other provinces of South Africa occurred from late October to late November 2021 (Viana et al., 2022). In these studies, selection and recombination analyses showed gene-wide positive selection and a possible single recombination event within the NTD region of the spike occurring after the appearance of the three known VOC omicron lineages. We note, however, that the maximum likelihood phylogenetic tree of Fig. 1A suggests the origin of VOC omicron is associated with an ancestor that appeared much earlier, perhaps dating back to mid-2020. Thus, earlier possible recombination events are still demanding dissection. Similarly, the rise of the complex molecular profile of VOC omicron and those of previous VOCs have not been explained. For example, the S-protein profile of VOC omicron in Australia (Fig. 7) follows the 37 mutations described by Viana et al., (2022) for the genomes of South Africa. The match, however, does not address the very early and noisy appearance of the individual mutations that make up the mutant constellation of VOC omicron in the different regions of Australia as it was displacing the VOC delta constellation in the last month of the last calendar quarter of 2021 (Figs. S1–S4 in Supplementary information). Note that the first reports of VOC omicron in Australia occurred on November 28–29, 2021, yet prevalence of sequences and daily cases increased massively some few weeks later (Fig. 1B) without reopening Australia's international and regional borders and under lockdowns, extensive contact tracing, and strong mandatory quarantine restrictions. How could this massive increase in prevalence occur in a country with one of the highest vaccination rates and toughest restriction policies of the planet? While some restrictions were lifted by the end of December 2021 following Phase 3 of the National Transition plan, our plots reveal VOC omicron was already overtaking the entire Australian viral pool. Was VOC omicron already present in significant numbers in Australia before the first infection cases were reported in South Africa?
An absence of clear evolutionary paths of transmission that could explain the origin of VOCs suggest an “emergence” in mutational landscapes of viral evolution. Here we explore this possible theoretical scenario. To do so, we studied patterns of mutation accumulation in regions of Australia, keeping only mutations that fulfill a “relevance” filtering criterion of significant presence in the evolving viral population. The mutations we identified and tracked matched profiles for the most prevalent VOCs that appeared in Australia (Fig. 1B), i.e., VOCs alpha, delta and omicron (Figs. S1–S4 in Supplementary information). We find that patterns of mutation accumulation were remarkably conserved for sets of mutations, a hallmark of haplotypes. This allowed to partition mutant constellations into haplotype groups and free-standing markers of VOCs, which were then studied by construction of haplotype networks.
7.1: Mutational landscapes of viral evolution
The SARS-CoV-2 genome is regarded as one of the most stable among positive-strand RNA viruses due to its NSP14-mediated 3′-5′ exoribonuclease proofreading activities, which repair polymerase errors during RNA replication (Ogando et al., 2020). However, the appearance of numerous variants, some of which jeopardize vaccination performance and many of which coincide with convergently gained spike mutations, has casted doubt on this notion of stability (Williams & Burgers, 2021). In fact, copying errors during viral replication combined with recombination, genomic re-assortment, and host-induced editing (e.g., via host RNA deaminases) push RNA viruses close to an “error threshold” of too many deleterious mutations compromising their persistence (Domingo, Sheldon, & Perales, 2012). When mutations of the “master” sequence combine with each other in the evolving viral population, this emerging “cloud” of variants defines a mutational landscape that the viral “quasispecies” seeks to optimize (Caetano-Anollés et al., 2022). In doing so, the viral population becomes “structured” by mutation accumulation. This structuring has been recently documented in the United States (Tasakis et al., 2021) and England (Vöhringer et al., 2021).
Tasakis et al. (2021) examined 62,211 SARS-CoV-2 genomes sampled in the United States from January 2020 through April 2021. The frequency of mutations were compared between variants in 42 states, as well as those imported from other countries. The study found mutations were accumulating in genomes and mutant strains were converting to VOCs through a combination of genetic drift and selection mediated by serial “super spreader” founder events in a sea of mutational bursts. Unique variants circulating in year 2020 were divided into two groups, lineages closely related to the parental Wuhan strain containing few variations, and descendants of the European G-clade containing the D614G mutation of the spike. Such variants likely arose due to single base substitutions that led to serial founder events. The virus continued to evolve by accumulating mutations even after implementation of public-wide vaccination in 2021. Mutational signature analysis also revealed an increasing trend of amino acid mutations, which suggested a primary role of RNA modifying enzymes in the generation of mutations. Fourteen of the most notable missense mutations (mostly C > T) with a frequency of more than 10% were under positive selection regimes (including T85I of NSP2, P323L of NSP12, D614G of the S-protein and Q57H of the ORF3a viroporin). Finally, the gradual appearance of variants between late 2020 and late March 2021, including more evolved strains of VOC alpha, highlighted the need for a combination of strict containment as well as vaccination approaches.
An epidemiological study in England conducted on 281,178 viral genome sequences collected from COVID-positive patients identified 328 PANGO lineages distributed in 315 English local authorities between September 2020 and June 2021 (Vöhringer et al., 2021). The highly-diverse lineages that were present in the Fall of 2020 were followed by the massive sweeps of VOCs alpha and delta. Remarkably, there was an observed pattern of sub-epidemic waves, each driven by new mutations, especially in the S-protein. For example, after the initial emergence of the virus, the second wave was predominantly led by two sublineages, B.1 and B.1.1, and interestingly, its prevalence pattern was geographically diverse with higher rates of prevalence observed in the North of England compared to the South. Similarly, the third wave was driven by VOC alpha. With every new epidemic wave, a new lineage and mutations came to the forefront, which helped the virus escape previous immunity. A number of refractory variants containing the E484K mutation of the S-protein (including VOCs beta, gamma, eta, iota and zeta) followed the demise of VOC alpha and preceded the rise of VOCs kappa and delta. These lineages introduced a number of new mutations, which were then replaced by the VOC delta constellation.
Both studies suggest that the SARS-CoV-2 genome is evolving dynamically, with bursts of mutation accumulation followed by sweeps. Patterns of sub-epidemic waves and the rise of VOCs suggest major global shifts in the selective landscape and possible convergence between lineages (Martin et al., 2022). In addition, the viral genome appears to undergo evolutionary change in response to its host, even under implementation of selective pressures such as vaccination. Mutational changes are particularly exacerbated when super spreader events drive infrequent mutations to prominence (Choi et al., 2020).
Our analysis of SARS-CoV-2 genomes in Australia is in line with findings for the United States and England. Mutations accumulated in bursts while haplotypes were generated in waves. Two haplotypes were the first to accumulate during the initial wave of the pandemic early in 2020, haplotype 5 (involving the S-protein and NSP12) and haplotype 2 (involving the N-protein). The rise of VOC alpha early in 2021 took advantage of these two haplotypes but also generated an additional three, haplotype 1 (involving the S-protein, N-protein, ORF8 and NSP3), haplotype 3 (involving the S-protein) and haplotype 4 (involving NSP6). The rise of VOC delta mid-2021 displaced most markers of VOC alpha but retained haplotype 5. Instead it generated 6 additional haplotypes of its own, which were highly enriched in markers of the S-protein. Remarkably, the arrival of VOC omicron at the end of 2021 displaced most markers from VOC delta but retained haplotype 5, recruited the vanished haplotypes 2, 3 and 4 and two free-standing S-protein markers of VOC alpha, and 3 S-protein markers and one NSP4 marker from VOC delta. The three successive waves involved 24, 35 and 59 markers, showcasing the gradual structuring of the mutational landscape of the viral population.
7.2: VOC emergence by haplotype coalescence
Our survey explores the rise of mutant constellations in Australia. We find constellations are made up of haplotypes and free-standing markers. The existence of an haplotype implies for example that evolutionary constraints are acting on intramolecular and intermolecular interactions. Collectively, these constraints are impacting the physiologies of the viral life cycle. Mutation accumulation plots show that many of these interactions do not rise monolithically. Instead, there is significant variation in the timing of appearance and accumulation of haplotype markers in the different regions of Australia. This suggests there is a global emergence process that depends on the regional environment and is developing independently of selective sweeps. To illustrate, Fig. 3A shows the noisy appearance of the two amino acid variants of haplotype 5, the oldest and most stable haplotype. Markers did not accumulate uniformly in the different regions as their prevalence was approaching 100%. They did so idiosyncratically as shown by accumulation curves for the two N-protein markers of haplotype 2 (Fig. 3B) Their fate was more haphazard and never reached 100% prevalence levels in regions closer to the Equator. In contrast, other haplotypes did not decouple but showed distinct accumulation patterns in the different regions (e.g., haplotype 1 of VOC alpha; Fig. 4A).
The atomization of mutant constellations into haplotypes was unanticipated and shows VOCs are highly dynamic entities that are capable of adding and eliminating markers from their individual makeup. However, there is some structure to the assembly of haplotypes. When there is no recombination or rearrangement, haplotype structure reflects the age distribution of mutations in an evolving phylogenetic tree of viral variants. In the presence of horizontal processes of genetic exchange, every sequence site that is subject to mutation can follow a tree but site-specific trees must be reconciled. In other words, horizontal exchange shuffles genetic variation. This complicates standard views of population genetics, such as forward-in-time evolution of allele frequencies and backward-in-time genealogical models. One example is the duality between the Wright-Fisher diffusion for genetic drift and its genealogical counterpart, the coalescent, which has been only recently modeled to incorporate the effects of recombination (Griffiths, Jenkins, & Lessard, 2016). Here, an ancestral recombination graph must be reconciled with genetic drift by diffusion in a landscape of mutations in an attempt to explain how ancestral genetic material is dispersed across ancestors of a contemporary population. In the case of VOCs of a viral quasispecies, we have little understanding of these processes. Instead, we can define haplotypes experimentally by detecting congruent patterns of accumulation of their markers. Patterns that are similar suggest a “coalescence” of haplotypes or free-standing markers into a same regional behavior. We exploit this coalescence in an “haplotype graph” that describes how haplotypes share markers and similarities in patterns of accumulation across regions of Australia (Fig. 5). Three clear subgraphs describing the three major VOCs that appeared in Australia are unified by the universally present haplotype 5 that contains the D614G marker of the spike. Other haplotypes and free-standing markers also unify VOC makeup. Haplotype coalescence is described by core haplotypes acting as hubs.
7.3: Haplotypes and seasonal behavior
Beta-coronaviruses, including SARS-CoV-2, are considered “winter viruses” because of their high transmission rates in the winter season. Winter viruses include the influenza virus (Tamerius et al., 2011), norovirus (Martinez, 2018), and common cold viruses (Eccles, 2005), all of which display the highest incidence rates during wintertime (Nickbakhsh et al., 2020). Transmission patterns in these seasonal viruses depend strongly on environmental factors, host susceptibility, and the type and level of immune response that the host mounts against viral infection. Direct and indirect contact play a significant role in transmission of respiratory viruses. Airborne transmission for example is greatly affected by susceptibility, weather, temperature, topography, and air quality, all of which contribute to seasonal behavior (Pica & Bouvier, 2012). In addition, the host's environment (e.g., time spent indoors), host defense (e.g., impaired mucociliary clearance through cold and dry air inhalation), and changes in viral infectability and stability along with climatic conditions are known to impact the seasonal behavior of respiratory viruses (Tamerius et al., 2011). Viral transmissibility can also be very high during initial stages of an epidemic due to lower immunity, and environmental factors cannot stop transmission at times (Grenfell & Bjørnstad, 2005). In the case of the influenza virus, temperature and relative humidity have the greatest impact on seasonality (Lowen, Mubareka, Steel, & Palese, 2007; Pica & Bouvier, 2012).
The effect of seasonal variations on the transmissibility of SARS-CoV-2 has become a topic of great interest (Kissler, Tedijanto, Goldstein, Grad, & Lipsitch, 2020). Seasonality warrants attention because it could assist in formulating informed actionable responses to the COVID-19 pandemic. Emerging evidence suggests that an association exists between seasonal variations and the survival and transmissibility of SARS-CoV-2, with higher latitude, colder temperatures, and lower humidity levels being associated with higher incidence of COVID-19 (Burra et al., 2021; Liu et al., 2021; Sajadi et al., 2020). Considering the structure of SARS-CoV-2, it is logical that the virus could be sensitive to environmental conditions. The viral capsid that encases the proteins and genetic material is surrounded by a lipid bilayer likely to be affected by environmental conditions like temperature and humidity (Moriyama, Hugentobler, & Iwasaki, 2020). In fact, our research suggests that the seasonality of COVID-19 can be explained by the presence of a galectin-like structure in the NTP region of the S1 subunit of the spike protein (Caetano-Anollés et al., 2022). The NTD, together with the RBD region, facilitates the viral attachment to cells by recognizing and binding to sugars and other receptors of the host cell (Pourrajab, 2021). Galectins are a family of evolutionarily conserved effector proteins that regulate a wide range of biological processes including pre-mRNA splicing and various kinds of cellular interactions, as well as pathogen recognition and inflammatory responses (Dings, Miller, Griffin, & Mayo, 2018). These diverse roles appear to mostly involve binding to the carbohydrate moieties of glycoconjugates present on the surface of cells (Caetano-Anollés et al., 2022). The presence of cellular structures similar to galectins in the viral spike protein suggests they play a role in helping SARS-CoV-2 attach to host cells by using the same carbohydrate receptors used by galectins when evading host immune responses (possibly by masking itself from host galectins; Pourrajab, 2021). Such hypotheses about the role of these galectin like structures in SARS-CoV-2 have been corroborated by findings on the administration of SARS-Cov-2 galectin like inhibitors, which reportedly decrease SARS-CoV-2 loads in patients (Sethi, Sanam, Munagalasetty, Jayanthi, & Alvala, 2020). Remarkably, a relationship between temperature shifts and the activity of galectins in clearance of pathogens of “cauliflower” corals (Pocillopora damicornis), which is necessary to establish a healthy symbiosis with dinoflagellates, was recently established (Wu et al., 2019). Lower temperatures were associated with increased pathogen survival. On the other hand, increased temperatures enhanced host cells galectin-mediated pathogen recognition and clearance with the activity reaching a maximum at the temperatures between 25 °C and 30 °C. We found a similar association between galectin activity and temperature in SARS-CoV-2 (Caetano-Anollés et al., 2022). It appears galectin-like structures significantly impact the seasonal behavior of SARS-CoV-2 and need to be further investigated.
The spike appears not the only protein mediating seasonal behavior in SARS-CoV-2. Our analysis revealed latitude-dependent differences in mutation accumulation patterns of haplotypes and free-standing markers. These differences involved markers affecting a wide range of proteins. When focusing on core haplotypes of VOCs, we detected significant diversity of accumulation patterns below a − 34°S latitude transect and highly coupled patterns at higher (and colder) latitudes of Australia (Fig. 8). The core haplotype of VOC alpha involved 4 proteins, the 6 haplotypes of VOC delta involved 12 proteins, and the 6 haplotypes of the core VOC omicron involved 7 proteins, all of which included a range of structural, accessory and non-structural proteins. Similarly, a focus on free-standing markers shared by VOCs revealed noisy patterns of accumulation suggestive of latitude-dependent recruitment of markers (Fig. 9). As with haplotypes, free-standing markers involved a range of 9 structural, accessory and non-structural proteins. While the S-protein was part of these mutant constellations, VOC alpha was enriched in mutants affecting regions that modulate flexibility/rigidity of the spike (including mutations in the C-terminal S2 subunit and the furin site), VOC delta was enriched in mutants altering the environment-sensing NTD region but also contained markers in the immunogenic RBD region, and now, VOC omicron is recruiting mutations in the RBD region necessary for immune evasion but at the same time balanced by mutations in the NTD and the S2 subunit regions. This progression mirrors the three successive phases of the pandemic we previously proposed: flexibility/rigidity, environmental sensing, and vaccine-driven immune escape (Caetano-Anollés et al., 2022).
7.4: Conclusions
Recent analyses of accumulating genome sequence data suggest that the SARS-CoV-2 virus is evolving in bursts while developing with each new VOC an increasing constellation of mutations. This indicates that the virus is furthering persistence by adapting to the external environment and to the physiology and behavior of its human hosts (Caetano-Anollés et al., 2022). Although most of the mutations are evolutionarily neutral, VOC markers are under strong positive selection and their haplotype structure suggest this is occurring by fostering beneficial intramolecular and/or intermolecular interactions. A rate of ~ 25 amino acid substitutions per year has been reported (estimated with a molecular clock) and the latest VOC omicron shows at least 37 mutations in the spike protein alone (Cameroni et al., 2021; Vöhringer et al., 2021). The increase in immune host populations by vaccination and treatments may lead to increasing immune evasion strategies. This is already evident in the large number of mutants targeting the immunogenic regions of the spike protein (Fig. 7). This highlights the need for genomic surveillance in different geographic locations of the world to better understand viral adaptation, mutational patterns, the rise of viral sub-lineages, and transmission.
Our study finds there is a rationale behind the emergence of VOCs in Australia. The noisy rise of haplotypes by molecular optimization involves a coalescence into monolithic constellations that are only decoupled by seasonal behavior. Thus, viral evolution is tailored by the seasonal periodicities of the planet that arise from Earth's tilted axis relative to the plane of its orbit. We will soon expect an endemic COVID-19 with outbreaks moving across the Earth every year along a sinuous curve parallel to the “midsummer” curve of solar radiation. Such behavior will follow patterns not far away from those of influenza (Deyle, Maher, Hernandez, Basu, & Sugihara, 2016; Hope-Simpson, 1981).
References
Artika, Dewantari and Wiyatno, 2020 Artika I.M., Dewantari A.K., Wiyatno A. Molecular biology of coronaviruses: Current knowledge. Heliyon. 2020;6(8):e04743.
Bai, Cao, Liu and Li, 2021 Bai Z., Cao Y., Liu W., Li J. The SARS-CoV-2 nucleocapsidprotein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses. 2021;13:1115.
Becerra-Flores and Cardozo, 2020 Becerra-Flores M., Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. International Journal of Clinical Practice. 2020;74:e13525.
Burra et al., 2021 Burra P., Soto-Díaz K., Chalen I., Gonzalez-Ricon R.J., Istanto D., Caetano-Anollés G. Temperature and latitude correlate with SARS-CoV-2 epidemiological variables but not with genomic change worldwide. Evolutionary Bioinformatics. 2021;19: 1176934321989695.
Caetano-Anollés, Hernandez, Mughal, Tomaszewski and Caetano-Anollés, 2022 Caetano-Anollés K., Hernandez N., Mughal F., Tomaszewski T., Caetano-Anollés G. The seasonal behaviour of COVID-19 and its galectin-like culprit of the viral spike. Methods in Microbiology. 2022. ;50:27–81. https://doi.org/10.1016/bs.mim.2021.10.002.
Cameroni et al., 2021 Cameroni E., Bowen J.E., Rosen L.E., Saliba C., Zepeda S.K., et al. Broadly neutralizing antibodies overcome SARS-CoV-2 omicron antigenic shift. Nature. 2021;602:664–670.
Campbell et al., 2021 Campbell F., Archer B., Laurenson-Schafer H., Jinnai Y., Konings F., et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at 2021. Euro Surveillance. 2021;26(24):2100509.
Caswell, Droettboom, Lee, et al., 2021 Caswell T.A., Droettboom M., Lee A., et al. Matplotlib/Matplotlib, v3.5.1. Zenodo. 2021;doi:10.5281/zenodo.5773480.
Choi et al., 2020 Choi B., Choudhary M.C., Regan J., Sparks J.A., Padera R.F., et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. New England Journal of Medicine. 2020;383(23):2291–2293.
Cottam et al., 2011 Cottam E.M., Maier H.J., Manifava M., Vaux L.C., Chandra-Schoenfelder P., et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7:1335–1347.
Davies et al., 2021 Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372 (6538) eabg3055.
Deyle, Maher, Hernandez, Basu and Sugihara, 2016 Deyle E.R., Maher M.C., Hernandez R.D., Basu S., Sugihara G. Global environmental drivers of influenza. Proceedings of the National Academy of Science of the USA. 2016;113 :13081–13086.
Dings, Miller, Griffin and Mayo, 2018 Dings R.P., Miller M.C., Griffin R.J., Mayo K.H. Galectins as molecular targets for therapeutic intervention. International Journal of Molecular Sciences. 2018;19(3):905.
Domingo, Sheldon and Perales, 2012 Domingo E., Sheldon J., Perales C. Viral quasispecies evolution. Microbiology and Molecular Biology Reviews. 2012;76:159–216.
Eccles, 2005 Eccles R. Understanding the symptoms of the common cold and influenza. The Lancet Infectious Diseases. 2005;5(11):718–725.
Elbe and Buckland-Merrett, 2017 Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Global Challenges. 2017;1:33–46.
Grenfell and Bjørnstad, 2005 Grenfell B., Bjørnstad O. Epidemic cycling and immunity. Nature. 2005;433(7024):366–367.
Griffiths, Jenkins and Lessard, 2016 Griffiths R.C., Jenkins P.A., Lessard S. A coalescent dual processes for a Wright-fisher diffusion with recombination and its application to haplotype partitioning. Theoretical Population Biology. 2016;112:126–138.
Harvey et al., 2021 Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., et al. SARS-CoV-2 variants, spike mutations and immune escape. Nature Reviews Microbiology. 2021;19:409–424.
Hodcroft, Hadfield, Neher and Bedford, 2020 Hodcroft E.B., Hadfield J., Neher R.A., Bedford T. Year-letter genetic clade naming for SARS-CoV-2 on Nextstrain.org. Nextstrain. 2020. https://nextstrain.org/blog/2020-06-02-SARSCoV2-clade-naming.
Hope-Simpson, 1981 Hope-Simpson R.E. The role of season in the epidemiology of influenza. Journal of Hygiene (London). 1981;86(1):35–47.
Hsieh, Li, Chen and Lo, 2008 Hsieh Y.-C., Li H.-C., Chen S.-C., Lo S.-Y. Interactions between M protein and other structural proteins of severe, acute respiratory syndrome-associated coronavirus. Journal of Biomedical Science. 2008;15(6):707–717.
Hunter, 2007 Hunter J.D. Matplotlib: A 2D graphics environment. Computing in Science & Engineering. 2007;9(3):90–95.
Hurst, Koetzner and Masters, 2013 Hurst K.R., Koetzner C.A., Masters P.S. Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. Journal of Virology. 2013;87:9159–9172.
Khare et al., 2021 Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., et al. GISAID's role in pandemic response. CCDC Weekly. 2021;3(49):1049–1051.
Kissler, Tedijanto, Goldstein, Grad and Lipsitch, 2020 Kissler S.M., Tedijanto C., Goldstein E., Grad Y.H., Lipsitch M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science. 2020;368(6493):860–868.
Korber et al., 2020 Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020;182:812–827.
Kumar, Kumar, Garg and Giri, 2021 Kumar P., Kumar A., Garg N., Giri R. An insight into SARS-CoV-2 membrane protein interaction with spike, envelope, and nucleocapsid proteins. Journal of Biomolecular Structure and Dynamics. 2021;doi:10.1080/07391102.2021.2016490.
Lauring and Hodcroft, 2021 Lauring A.S., Hodcroft E.B. Genetic variants of SARS-CoV-2—What do they mean?. JAMA. 2021;325(6):529–531. doi:10.1001/jama.2020.27124.
Lei, Kusov and Hilgenfeld, 2018 Lei J., Kusov Y., Hilgenfeld R. NSP3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Research. 2018;149:58–74.
Liu et al., 2021 Liu X., Huang J., Li C., Zhao Y., Wang D., Huang Z., et al. The role of seasonality in the spread of COVID-19 pandemic. Environmental Research. 2021;195:110874.
Lowen, Mubareka, Steel and Palese, 2007 Lowen A.C., Mubareka S., Steel J., Palese P. Influenza virus transmission is dependent on relative humidity and temperature. PLoS Pathogens. 2007;3(10):e151.
Lu et al., 2021 Lu S., Ye Q., Singh D., Cao Y., Diedrich J.K., et al. The SARS-CoV-2 Nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nature Communications. 2021;12:502.
Lutomski, El-Baba, Bolla and Robinson, 2021 Lutomski C.A., El-Baba T.J., Bolla J.R., Robinson C.V. Multiple roles of SARS-CoV-2 N protein facilitated by proteoform-specific interactions with RNA, host proteins, and convalescent antibodies. JACS Au. 2021;1:1147–1157.
Mallapaty, 2022 Mallapaty S. The hunt for the origins of omicron. Nature. 2022;602:26–27.
Martin et al., 2022 Martin D.P., Lytras S., Lucaci A.G., Maier W., Grüning B., et al. Selection analysis identifies unsusual clustered mutational changes in omicron lineage BA.1 that likely impact spike function. bioRxiv. 2022. https://www.biorxiv.org/content/10.1101/2022.01.14.476382v1.
Martinez, 2018 Martinez M.E. The calendar of epidemics: Seasonal cycles of infectious diseases. PLoS Pathogens. 2018;14(11):e1007327.
McKinney, 2010 McKinney W. Data structures for statistical computing in Python. In: van der Walt S., Millman J., eds. Proceedings of the 9th Python in science conference. 2010:56–61.
Moriyama, Hugentobler and Iwasaki, 2020 Moriyama M., Hugentobler W.J., Iwasaki A. Seasonality of respiratory viral infections. Annual Review of Virology. 2020;7:83–101.
Nickbakhsh et al., 2020 Nickbakhsh S., Ho A., Marques D.F., McMenamin J., Gunson R.N., Murcia P.R. Epidemiology of seasonal coronaviruses: Establishing the context for the emergence of coronavirus disease 2019. The Journal of Infectious Diseases. 2020;222(1):17–25.
Ogando et al., 2020 Ogando N.S., Zevenhoven-Dobbe J.C., Meer Y.V.D., Bredenbeek P.J., Posthuma C.C., Snijder E.J., et al. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. Journal of Virology. 2020;94(23) e01246–01220.
Pandas Development Team, 2022 Pandas Development Team. Pandas-dev/pandas: Pandas 1.1.5. Zenodo. 2022;doi:10.5281/zenodo.5893288.
Pica and Bouvier, 2012 Pica N., Bouvier N.M. Environmental factors affecting the transmission of respiratory viruses. Current Opinion in Virology. 2012;2(1):90–95.
Pourrajab, 2021 Pourrajab F. Targeting the glycans: A paradigm for host-targeted and COVID-19 drug design. Journal of Cellular and Molecular Medicine. 2021;25(13):5842–5856.
Rambault et al., 2020 Rambault A., Holmes E.C., O'Toole A., Hill V., McCrone J.T., Ruis C., et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nature Microbiology. 2020;5:1403–1407.
Rodrigues et al., 2021 Rodrigues T.S., de Sá K.S.G., Ishimoto A.Y., Becerra A., Oliveira S., et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. Journal of Experimental Medicine. 2021;218:e20201707.
Sajadi et al., 2020 Sajadi M.M., Habibzadeh P., Vintzileos A., Shokouhi S., Miralles-Wilhelm F., Amoroso A. Temperature, humidity, and latitude analysis to estimate potential spread and seasonality of coronavirus disease 2019 (COVID-19). JAMA Network Open. 2020;3:e2011834.
Sethi, Sanam, Munagalasetty, Jayanthi and Alvala, 2020 Sethi A., Sanam S., Munagalasetty S., Jayanthi S., Alvala M. Understanding the role of galectin inhibitors as potential candidates for SARS-CoV-2 spike protein: In silico studies. RSC Advances. 2020;10(50):29873–29884.
Showers, Leach, Kechris and Strong, 2022 Showers W.M., Leach S.M., Kechris K., Strong M. Analysis of SARS-CoV-2 mutations over time reveals increasing prevalence of variants in the spike protein and RNA-dependent RNA polymerase. Infection, Genetics and Evolution. 2022;97:105153.
Shu and McCauley, 2017 Shu Y., McCauley J. GISAID: Global initiative on sharing all influenza data – From vision to reality. Eurosurveillance. 2017;22(13):30494.
Starr et al., 2020 Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;5:1295–1310.
Sun, Huang, Xu, & Hu, 2021 Sun X., Huang Z., Xu W., Hu W., et al. SARS-CoV-2 non-strucural protein 6 triggers LRRP3-dependent pyroptosis by targeting TP6AP1. Cell Death & Differentiation. 2021;doi:10.1038/s41418-021-00916-7.
Tamerius et al., 2011 Tamerius J., Nelson M.I., Zhou S.Z., Viboud C., Miller M.A., Alonso W.J. Global influenza seasonality: Reconciling patterns across temperate and tropical regions. Environmental Health Perspectives. 2011;119(4):439–445.
Tasakis et al., 2021 Tasakis R.N., Samaras G., Jamison A., Lee M., Paulus A., et al. SARS-CoV-2 variant evolution in the United States: High accumulation of viral mutations over time likely through serial founder events and mutational bursts. bioRxiv. 2021;doi:10.1371/journal.pone.0255169.
Tomaszewski et al., 2020 Tomaszewski T., DeVriers R.S., Dong M., Bhatia G., Norsworthy M.D., Zheng X., et al. New pathways of mutational change in SARS-CoV-2 proteomes involve regions of intrinsic disorder important for virus replication and release. Evolutionary Bioinformatics. 2020;16: 1176934320965149.
Viana et al., 2022 Viana R., Moyo S., Amoako D.G., Tegally H., Scheepers C., et al. Rapid epidemic expansion of the SARS-CoV-2 omicron variant in southern Africa. Nature. 2022. https://doi.org/10.1038/s41586-022-04411-y.
Vöhringer et al., 2021 Vöhringer H.S., Sanderson T., Sinnott M., De Maio N., Nguyen T., et al. Genomic reconstruction of the SARS-CoV-2 epidemic in England. Nature. 2021;600(7889):506–511.
Voltz et al., 2021 Voltz E., Hill V., McCrone J.T., Proce A., Jorgensen D., et al. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogencity. Cell. 2021;184(1):64–75.
Williams and Burgers, 2021 Williams T.C., Burgers W.A. SARS-CoV-2 evolution and vaccines: Cause for concern?. The Lancet Respiratory Medicine. 2021;9(4):333–335.
Wu et al., 2019 Wu Y., Zhou Z., Wang J., Luo J., Wang L., Zhang Y. Temperature regulates the recognition activities of a galectin to pathogen and symbiont in the scleractinian coral Pocillopora damicornis. Developmental & Comparative Immunology. 2019;96:103–110.
Xu et al., 2021 Xu C., Wang Y., Liu C., Zhang C., Han W., Hong X., et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Science Advances. 2021;7 (1) eabe5575.
Zhang et al., 2021 Zhang Y., Sun H., Pei R., Mao B., Zhao Z., Li H., et al. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discovery. 2021;7:31.