5
“The great tragedy of science—the slaying of a
beautiful hypothesis by an ugly fact.”
—THOMAS HUXLEY (1870)1
The adult human brain weighs about three pounds, and when you see it close up, removed from the skull, it is a bit larger than you imagined it to be. I had thought a brain could rest fairly easily in the palm of one’s hand, but you really need both hands to lift it securely into the air. If the brain is fresh, not yet pickled in formaldehyde, a spiderweb of blood vessels pinkens the surface, and the tissue feels soft, almost gelatinous. It is definitely “biological” in kind, and yet somehow it gives rise to all of the mysterious and remarkable talents of the human mind. At the invitation of a friend, Jang-Ho Cha, who is a neuroscientist at Massachusetts General Hospital, I attended a brain-cutting seminar at the hospital, with the thought that seeing a human brain would help me better visualize the neurotransmitter pathways that are said to give rise to depression and psychosis, but naturally my visit turned into something more than that. The human brain up close takes your breath away.
The mechanics of its messaging system are fairly well understood. There are, Cha noted, 100 billion neurons in the human brain. The cell body of a “typical” neuron receives input from a vast web of dendrites, and it sends out a signal via a single axon that may project to a distant area of the brain (or down the spinal cord). At its end, an axon branches into numerous terminals, and it is from these terminals that chemical messengers—dopamine, serotonin, etc.—are released into the synaptic cleft, which is a gap about twenty nanometers wide (a nanometer is one-billionth of a meter). A single neuron has between one thousand and ten thousand synaptic connections, with the adult brain as a whole having perhaps 150 trillion synapses.
The axons of neurons that use the same neurotransmitter are regularly bundled together, almost like the strands of a telecommunications cable, and once scientists discovered that dopamine, norepinephrine, and serotonin fluoresced different colors when exposed to formaldehyde vapors, it became possible to track those neurotransmitter pathways in the brain. Although Joseph Schildkraut, when he formulated his theory of affective disorders, thought that norepinephrine was the neurotransmitter most likely to be in short supply in those who were depressed, researchers fairly quickly turned much of their attention to serotonin, and so for our purposes, in regard to our investigation of the chemical imbalance theory of mental disorders, we need to look at that pathway in the brain for depression, and at the dopaminergic pathway for schizophrenia.
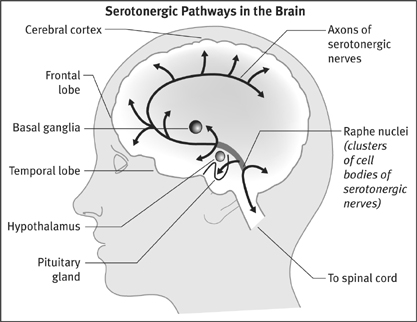
The serotonergic pathway is one with ancient evolutionary roots. Serotonergic neurons are found in the nervous systems of all vertebrates and most invertebrates, and in humans their cell bodies are located in the brain stem, in an area known as the raphe nuclei. Some of these neurons send long axons down the spinal cord, a system that is involved in the control of respiratory, cardiac, and gastrointestinal activities. Other serotonergic neurons have axons that ascend into all areas of the brain—the cerebellum, the hypothalamus, the basal ganglia, the temporal lobes, the limbic system, the cerebral cortex, and the frontal lobes. This pathway is involved in memory, learning, sleep, appetite, and the regulation of moods and behaviors. As Efrain Azmitia, a professor of biology at NYU, has noted, “the brain serotonin system is the single largest brain system known and can be characterized as a ‘giant’ neuronal system.”2
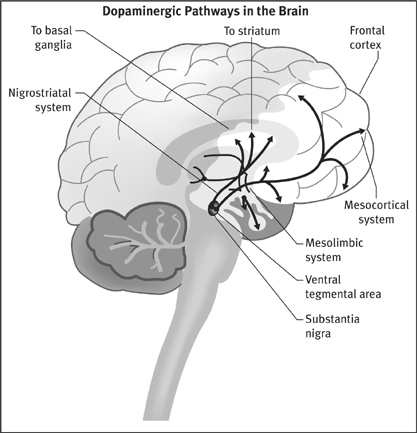
There are three major dopaminergic pathways in the brain. The cell bodies of all three systems are located atop the brain stem, in either the substantia nigra or the ventral tegmentum. Their axons project to the basal ganglia (nigrostriatal system), the limbic region (mesolimbic system), and the frontal lobes (mesocortical system). The basal ganglia initiates and controls movement. The limbic structures—the olfactory tubercle, the nucleus accumbens, and the amygdala, among others—are located behind the frontal lobes and help regulate our emotions. It is here that we feel the world, a process that is vital to our sense of self and our conceptions of reality. The frontal lobes are the most distinguishing feature of the human brain, and provide us with the godlike capacity to monitor our own selves.
All of this physiology—the 100 billion neurons, the 150 trillion synapses, the various neurotransmitter pathways—tell of a brain that is almost infinitely complex. Yet the chemical imbalance theory of mental disorders boiled this complexity down to a simple disease mechanism, one easy to grasp. In depression, the problem was that the serotonergic neurons released too little serotonin into the synaptic gap, and thus the serotonergic pathways in the brain were “underactive.” Antidepressants brought serotonin levels in the synaptic gap up to normal, and that allowed these pathways to transmit messages at a proper pace. Meanwhile, the hallucinations and voices that characterized schizophrenia resulted from overactive dopaminergic pathways. Either the presynaptic neurons pumped out too much dopamine into the synapse or the target neurons had an abnormally high density of dopamine receptors. Antipsychotics put a brake on this system, and this allowed the dopaminergic pathways to function in a more normal manner.
That was the chemical imbalance theory put forth by Schildkraut and Jacques Van Rossum, and the very research that had led Schildkraut to his hypothesis also provided investigators with a method for testing it. The studies of iproniazid and imipramine had shown that neurotransmitters were removed from the synapse in one of two ways. Either the chemical was taken back up into the presynaptic neuron and restored for later use, or it was metabolized by an enzyme and carted off as waste. Serotonin is metabolized into 5-hydroxyindole acetic acid (5-HIAA); dopamine is turned into homo vanillic acid (HVA). Researchers could comb the cerebrospinal fluid for these metabolites, and the amounts found would serve as an indirect gauge of the synaptic levels of the neurotransmitters. Since low serotonin was theorized to cause depression, anyone in that emotional state should have lower-than-normal levels of 5-HIAA in his or her cerebrospinal fluid. Similarly, since an overactive dopamine system was theorized to cause schizophrenia, people who heard voices or were paranoid should have abnormally high cerebrospinal levels of HVA.
This line of research kept scientists busy for nearly fifteen years.
The Serotonin Hypothesis Is Put to the Test
In 1969, Malcolm Bowers at Yale University became the first to report on whether depressed patients had low levels of serotonin metabolites in their cerebrospinal fluid. In a study of eight depressed patients (all of whom had been previously exposed to antidepressants), he announced that their 5-HIAA levels were lower than normal, but not “significantly” so.3 Two years later, investigators at McGill University said that they, too, had failed to find a “statistically significant” difference in the 5-HIAA levels of depressed patients and normal controls, and that they also had failed to find any correlation between 5-HIAA levels and the severity of depressive symptoms.4 In 1974, Bowers was back with a more finely tuned follow-up study: Depressed patients who had not been exposed to antidepressants had perfectly normal 5-HIAA levels.5
The serotonin theory of depression did not seem to be panning out, and in 1974, two researchers at the University of Pennsylvania, Joseph Mendels and Alan Frazer, revisited the evidence that had led Schildkraut to advance his theory in the first place. Schildkraut had noted that reserpine, which depleted monoamines in the brain (norepinephrine, serotonin, and dopamine), regularly made people depressed. But when Mendels and Frazer looked closely at the scientific literature, they found that when hypertensive patients were given reserpine, only 6 percent in fact got the blues. Furthermore, in 1955, a group of physicians in England had given the herbal drug to their depressed patients, and it had lifted the spirits of many. Reserpine, Mendels and Frazer concluded, didn’t reliably induce depression at all.6 They also noted that when researchers had given other monoamine-depleting drugs to people, those agents hadn’t induced depression either. “The literature reviewed here strongly suggests that the depletion of brain norepinephrine, dopamine or serotonin is in itself not sufficient to account for the development of the clinical syndrome of depression,” they wrote.7
It seemed that the theory was about to be declared dead and buried, but then, in 1975, Marie Asberg and her colleagues at the Karolinska Institute in Stockholm breathed new life into it. Twenty of the sixty-eight depressed patients they had tested suffered from low 5-HIAA levels, and these low-serotonin patients were somewhat more suicidal than the rest, with two of the twenty eventually committing suicide. This was evidence, the Swedish researchers said, that there might be “a biochemical subgroup of depressive disorder characterized by a disturbance of serotonin turnover.”8
Soon prominent psychiatrists in the United States were writing that “nearly 30 percent” of depressed patients had been found to have low serotonin levels. The serotonin theory of depression seemed at least partly vindicated. But today, if we revisit Asberg’s study and examine her data, we can see that her finding of a “biological subgroup” of depressed patients was mostly a story of wishful thinking.
In her study, Asberg reported that 25 percent of her “normal” group had cerebrospinal 5-HIAA levels below fifteen nanograms per milliliter. Fifty percent had fifteen to twenty-five nanograms of 5-HIAA per milliliter, and the remaining 25 percent had levels above twenty-five nanograms. The bell curve for her “normals” showed that 5-HIAA levels were quite variable. But what she failed to note in her discussion was that the bell curve for the sixty-eight depressed patients in her study was almost exactly the same. Twenty-nine percent (twenty of the sixty-eight) had 5-HIAA counts below fifteen nanograms, 47 percent had levels between fifteen and twenty-five nanograms, and 24 percent had levels above twenty-five nanograms. Twenty-nine percent of depressed patients may have had “low” levels of serotonin metabolites in their cerebrospinal fluid (this was her “biological subgroup”), but then so did 25 percent of “normal” people. The median level for normals was twenty nanograms, and, it so turned out, more than half of the depressed patients—thirty-seven of sixty-eight—had levels above that amount.
Viewed in this way, her study had not provided any new reason to believe in the serotonin theory of depression. Japanese investigators soon revealed, in an unwitting way, the faulty logic at work. They reported that some antidepressants (used in Japan) blocked serotonin receptors, inhibiting the firing of those pathways, and thus they reasoned that depression might be caused by an “excess of free serotonin in the synaptic cleft.”9 They had applied the same backwards reasoning that had given rise to the low-serotonin theory of depression, and if the Japanese scientists had wanted to, they could have pointed to Asberg’s study for support of their theory, as the Swedes had found that 24 percent of depressed patients had “high” levels of serotonin.
In 1984, NIMH investigators studied the low-serotonin theory of depression one more time. They wanted to see whether the “biological subgroup” of depressed patients with “low” levels of serotonin were the best responders to an antidepressant, amitriptyline, that selectively blocked its reuptake. If an antidepressant was an antidote to a chemical imbalance in the brain, then amitriptyline should be most effective in that subgroup. But, lead investigator James Maas wrote, “contrary to expectations, no relationships between cerebrospinal 5-HIAA and response to amitriptyline were found.”10 Moreover, he and the other NIMH researchers discovered—just as Asberg had—that 5-HIAA levels varied widely in depressed patients. Some had high levels of serotonin metabolites in their cerebrospinal fluid, while others had low levels. The NIMH scientists drew the only possible conclusion: “Elevations or decrements in the functioning of serotonergic systems per se are not likely to be associated with depression.”*
Even after this report, the serotonin theory of depression did not completely go away. The commercial success of Prozac, a “selective serotonin reuptake inhibitor” brought to market in 1988 by Eli Lilly, fueled a new round of public claims that depression was due to low levels of this neurotransmitter, and once again any number of investigators conducted experiments to see if that were so. But this second round of studies produced the same results as the first. “I spent the first several years of my career doing full-time research on brain serotonin metabolism, but I never saw any convincing evidence that any psychiatric disorder, including depression, results from a deficiency of brain serotonin,” said Stanford psychiatrist David Burns in 2003.11 Numerous others made this same point. “There is no scientific evidence whatsoever that clinical depression is due to any kind of biological deficit state,” wrote Colin Ross, an associate professor of psychiatry at Southwest Medical Center in Dallas, in his 1995 book, Pseudoscience in Biological Psychiatry.12 In 2000, the authors of Essential Psychopharmacology told medical students “there is no clear and convincing evidence that monoamine deficiency accounts for depression; that is, there is no ‘real’ monoamine deficit.”13 Yet, fueled by pharmaceutical advertisements, the belief lived on, and it caused Irish psychiatrist David Healy, who has written a number of books on the history of psychiatry, to quip in 2005 that this theory needed to be put into the medical dustbin, where other such discredited theories can be found. “The serotonin theory of depression,” he wrote, with evident exasperation, “is comparable to the masturbatory theory of insanity.”14
Dopamine Deja Vu
When Van Rossum set forth his dopamine hypothesis of schizophrenia, he noted that the first thing that investigators needed to do was “further substantiate” that antipsychotic drugs did indeed thwart dopamine transmission in the brain. This took some time, but by 1975, Solomon Snyder at Johns Hopkins Medical School and Philip Seeman at the University of Toronto had fleshed out how the drugs achieved that effect. First, Snyder identified two distinct types of dopamine receptors, known as D1 and D2. Next, both investigators found that antipsychotics blocked 70 to 90 percent of the D2 receptors.15 Newspapers now told of how these drugs might be correcting a chemical imbalance in the brain.
“Too much dopamine function in the brain could account for the overwhelming flood of sensations that plagues the schizophrenic,” the New York Times explained. “By blocking the brain’s receptor sites for dopamine, neuroleptics put an end to sights and sounds that are not really there.”16
However, even as Snyder and Seeman were reporting their results, Malcolm Bowers was announcing findings that cast a cloud over the dopamine hypothesis. He had measured the level of dopamine metabolites in the cerebrospinal fluid of unmedicated schizophrenics and found them to be quite normal. “Our findings,” he wrote, “do not furnish neurochemical evidence for an over-arousal in these patients emanating from a midbrain dopamine system.”17 Others soon reported similar results. In 1975, Robert Post at the NIMH determined that HVA levels in the cerebrospinal fluid of twenty unmedicated schizophrenics “were not significantly different from controls.”18 Autopsy studies also revealed that the brain tissue of drug-free schizophrenics did not have abnormal levels of dopamine. In 1982, UCLA’s John Haracz reviewed this body of research and drew the obvious bottom-line conclusion: “These findings do not support the presence of elevated dopamine turnover in the brains of [unmedicated] schizophrenics.”19
Having discovered that dopamine levels in never-medicated schizophrenics were normal, researchers turned their attention to a second possibility. Perhaps people with schizophrenia had an overabundance of dopamine receptors. If so, the postsynaptic neurons would be “hypersensitive” to dopamine, and this would cause the dopaminergic pathways to be overstimulated. In 1978, Philip See-man at the University of Toronto announced in Nature that this was indeed the case. At autopsy, the brains of twenty schizophrenics had 70 percent more D2 receptors than normal. At first glance, it seemed that the cause of schizophrenia had been found, but Seeman cautioned that all of the patients had been on neuroleptics prior to their deaths. “Although these results are apparently compatible with the dopamine hypothesis of schizophrenia in general,” he wrote, the increase in D2 receptors might “have resulted from the long-term administration of neuroleptics.”20
A variety of studies quickly proved that the drugs were indeed the culprit. When rats were fed neuroleptics, their D2 receptors quickly increased in number.21 If rats were given a drug that blocked D1 receptors, that receptor subtype increased in density.22 In each instance, the increase was evidence of the brain trying to compensate for the drug’s blocking of its signals. Then, in 1982, Angus MacKay and his British colleagues reported that when they examined brain tissue from forty-eight deceased schizophrenics, “the increases in [D2] receptors were seen only in patients in whom neuroleptic medication had been maintained until the time of death, indicating that they were entirely iatrogenic [drug-caused].”23 A few years later, German investigators reported the same results from their autopsy studies.24 Finally, investigators in France, Sweden, and Finland used positron emission topography to study D2-receptor densities in living patients who had never been exposed to neuroleptics, and all reported “no significant differences” between the schizophrenics and “normal controls.”25
Since that time, researchers have continued to study whether there might be something amiss with the dopaminergic pathways in people diagnosed with schizophrenia, and now and then someone reports having found an abnormality of some type in a subset of patients. But by the end of the 1980s, it was clear that the chemical-imbalance hypothesis of schizophrenia—that this was a disease characterized by a hyperactive dopamine system that was then put somewhat back into balance by the drugs—had come to a crashing end. “The dopaminergic theory of schizophrenia retains little credibility for psychiatrists,” observed Pierre Deniker in 1990.26 Four years later, John Kane, a well-known psychiatrist at Long Island Jewish Medical Center, echoed the sentiment, noting that there was “no good evidence for any perturbation of the dopamine function in schizophrenia.”27 Still, the public continued to be told that people diagnosed with schizophrenia had overactive dopamine systems, with the drugs likened to “insulin for diabetes,” and thus former NIMH director Steve Hyman, in his 2002 book, Molecular Neuropharmacology, was moved to once again remind readers of the truth. “There is no compelling evidence that a lesion in the dopamine system is a primary cause of schizophrenia,” he wrote.28
Requiem for a Theory
The low-serotonin hypothesis of depression and the high-dopamine hypothesis of schizophrenia had always been the twin pillars of the chemical-imbalance theory of mental disorders, and by the late 1980s, both had been found wanting. Other mental disorders have also been touted to the public as diseases caused by chemical imbalances, but there was never any evidence to support those claims. Parents were told that children diagnosed with attention deficit hyperactivity disorder suffered from low dopamine levels, but the only reason they were told that was because Ritalin stirred neurons to release extra dopamine. This became the storytelling formula that was relied upon by pharmaceutical companies again and again: Researchers would identify the mechanism of action for a class of drugs, how the drugs either lowered or raised levels of a brain neurotransmitter, and soon the public would be told that people treated with those medications suffered from the opposite problem.
From a scientific point of view, it is apparent today that the chemical imbalance hypothesis was always wobbly in kind, and many of the scientists who watched its rise and fall have looked back on it with a bit of embarrassment. As early as 1975, Joseph Mendels and Alan Frazer had concluded that Schildkraut’s hy pothesis of depression had arisen out of “tunnel thinking” that relied on an “inadequate evaluation of certain findings not consistent with the initial assumption.”29 In 1990, Deniker said that the same was true of the dopamine hypothesis of schizophrenia. When psychiatric researchers recast the drugs as “antischizophrenic” agents, he noted, they had gone “a bit far … one can say that neuroleptics diminish certain phenomena of schizophrenia, but [the drugs] do not pretend to be an etiological treatment of these psychoses.”30 The chemical-imbalance theory of mental disorders, wrote David Healy, in his book The Creation of Psychopharmacology, was embraced by psychiatrists because it “set the stage” for them “to become real doctors.”31 Doctors in internal medicine had their antibiotics, and now psychiatrists could have their “anti-disease” pills too.
Yet a societal belief in chemical imbalances has remained (for reasons that will be explored later), and it has led those who have investigated and written about this history to emphasize, time and again, the same bottom-line conclusion. “The evidence does not support any of the biochemical theories of mental illness,” concluded Elliot Valenstein, a professor of neuroscience at the University of Michigan, in his 1998 book Blaming the Brain.32 Even U.S. surgeon general David Satcher, in his 1999 report Mental Health, confessed that “the precise causes [etiologies] of mental disorders are not known.”33 In Prozac Backlash, Joseph Glenmullen, an instructor of psychiatry at Harvard Medical School, noted that “in every instance where such an imbalance was thought to be found, it was later proved to be false.”34 Finally, in 2005, Kenneth Kendler, coeditor in chief of Psychological Medicine, penned an admirably succinct epitaph for this whole story: “We have hunted for big simple neurochemical explanations for psychiatric disorders and have not found them.”35
This brings us to our next big question: If psychiatric drugs don’t fix abnormal brain chemistry, what do they do?
Prozac on My Mind
During the 1970s and 1980s, investigators put together detailed accounts of how the various classes of psychiatric drugs act on the brain, and how the brain in turn reacts to the drugs. We could relate the history of antidepressants, neuroleptics, benzodiazepines, or stimulants, and all of those histories would tell of a somewhat common process at work. But since the story of chemical imbalances in the public mind really took off after Eli Lilly brought Prozac (fluoxetine) to market, it seems appropriate to review what Eli Lilly scientists and other investigators, in reports published in scientific journals, had to say about how this “selective serotonin reuptake inhibitor” actually worked.
As was noted earlier, once a presynaptic neuron has released serotonin into the synaptic gap, it must be quickly removed so that the signal can be crisply terminated. An enzyme metabolizes a small amount; the rest is pumped back into the presynaptic neuron, entering via a channel known as SERT (serotonin reuptake transport). Fluoxetine blocks this reuptake channel, and as a result, Eli Lilly scientist James Clemens wrote in 1975, it causes a “pile-up of serotonin at the synapse.”36
However, as the Eli Lilly investigators discovered, a feedback mechanism then kicks in. The presynaptic neuron has “auto-receptors” on its terminal membrane that monitor the level of serotonin in the synapse. If serotonin levels are too low, one scientist quipped, these autoreceptors scream “turn on the serotonin machine.” If serotonin levels are too high, they scream “turn it off.” This is a feedback loop designed by evolution to keep the serotonergic system in balance, and fluoxetine triggers the latter message. With serotonin no longer being whisked away from the synapse, the autoreceptors tell the presynaptic neurons to fire at a dramatically lower rate. They begin to release lower-than-normal amounts of serotonin into the synapse.
Feedback mechanisms also change the postsynaptic neurons. Within four weeks, the density of their serotonin receptors drops 25 percent below normal, Eli Lilly scientists reported in 1981.37 Other investigators subsequently reported that “chronic fluoxetine treatment” may lead to a 50 percent reduction in serotonin receptors in certain areas of the brain.38 As a result, the post synaptic neurons become “desensitized” to the chemical messenger.
At this point, it may seem that the brain has successfully adapted to the drug. Fluoxetine blocks the normal reuptake of serotonin from the synapse, but the presynaptic neurons then begin releasing less serotonin and the postsynaptic neurons become less sensitive to serotonin and thus don’t fire so readily. The drug was designed to accelerate the serotonergic pathway; the brain responded by putting on the brake. It has kept its serotonergic pathway more or less in balance, an adaptive response that researchers have dubbed “synaptic resilience.”39 However, there is one other change that occurs during this initial two-week period, and it ultimately short-circuits the brain’s compensatory response. The autoreceptors for serotonin on the presynaptic neurons decline in number. As a result, this feedback mechanism becomes partially disabled, and the “turn off the serotonin machine” message dims. The presynaptic neurons begin to fire at a normal rate again, at least for a while, and to release more serotonin than normal each time. 40 *
As the Eli Lilly scientists and others put together this picture of fluoxetine’s effects on the brain, they speculated as to what part of this process was responsible for the drug’s antidepressant properties. Psychiatrists had long observed that antidepressants took two or three weeks to “work,” and thus the Eli Lilly researchers reasoned in 1981 that it was the decline in serotonin receptors, which took several weeks to occur, that was “the underlying mechanism associated with the therapeutic response.”41 If so, the drug could be said to work because it drove the serotonergic system into a less responsive state. But once researchers discovered that fluoxetine partially disabled the feedback mechanism, Claude de Montigny at McGill University argued that this was what allowed the drug to begin working. This disabling process also took two or three weeks to occur, and it allowed the presynaptic neurons to begin pumping higher amounts of serotonin than normal into the synapse. At that point, with fluoxetine continuing to block serotonin’s removal, the neurotransmitter could now indeed “pile up” in the synapse, and that would lead “to an enhancement of central serotonergic neurotransmission,” de Montigny wrote.42
That is the scientific story of how fluoxetine alters the brain, and it may be that this process helps depressed people get well and stay well. Only the outcomes literature can reveal whether that is so. But the medicine clearly doesn’t fix a chemical imbalance in the brain. Instead, it does precisely the opposite. Prior to being medicated, a depressed person has no known chemical imbalance. Fluoxetine then gums up the normal removal of serotonin from the synapse, and that triggers a cascade of changes, and several weeks later the serotonergic pathway is operating in a decidedly abnormal manner. The presynaptic neuron is putting out more serotonin than usual. Its serotonin reuptake channels are blocked by the drug. The system’s feedback loop is partially disabled. The postsynaptic neurons are “desensitized” to serotonin. Mechanically speaking, the serotonergic system is now rather mucked up.
Eli Lilly’s scientists were well aware that this was so. In 1977, Ray Fuller and David Wong observed that fluoxetine, since it disrupted serotonergic pathways, could be used to study “the role of serotonin neurons in various brain functions—behavior, sleep, regulation of pituitary hormone release, thermoregulation, pain responsiveness and so on.” To conduct such experiments, researchers could administer fluoxetine to animals and observe which functions became compromised. They would look for pathologies to appear. This type of research in fact was already being done: Fuller and Wong reported in 1977 that the drug stirred “stereotyped hyperactivity” in rats and “suppressed REM sleep” in both rats and cats.43
In 1991, in a paper published in the Journal of Clinical Psychiatry, Princeton neuroscientist Barry Jacobs made this very point about SSRIs. He wrote:
These drugs “alter the level of synaptic transmission beyond the physiologic range achieved under [normal] environmental/biological conditions. Thus, any behavioral or physiologic change produced under these conditions might more appropriately be considered pathologic, rather than reflective of the normal biological role of 5-HT [serotonin.]”44
During the 1970s and 1980s, researchers studying the effects of neuroleptics fleshed out a similar story. Thorazine and other standard antipsychotics block 70 to 90 percent of all D2 receptors in the brain. In response, the presynaptic neurons begin pumping out more dopamine and the postsynaptic neurons increase the density of their D2 receptors by 30 percent or more. In this manner, the brain is trying to “compensate” for the drug’s effects so that it can maintain the transmission of messages along its dopaminergic pathways. However, after about three weeks, the pathway’s feedback mechanism begins to fail, and the presynaptic neurons begin to fire in irregular patterns or turn quiescent. It is this “inactivation” of dopaminergic pathways that “may be the basis for the antipsychotic action,” explains the American Psychiatric Association’s Textbook of Psychopharmacology.45
Once again, this is a story of neurotransmitter pathways that have been transformed by the medication. After several weeks, their feedback loops are partially disabled, the presynaptic neurons are releasing less dopamine than normal, the drug is thwarting dopamine’s effects by blocking D2receptors, and the postsynaptic neurons have an abnormally high density of these receptors. The drugs do not normalize brain chemistry, but disturb it, and if Jacob’s reasoning is followed, to a degree that could be considered “pathological.”
A Paradigm for Understanding Psychotropic Drugs
Today, as provost of Harvard University, Steve Hyman is mostly engaged in the many political and administrative tasks that come with leading a large institution. But he is a neuroscientist by training, and in 1996 to 2001, when he was the director of the NIMH, he wrote a paper, one both memorable and provocative in kind, that summed up all that had been learned about psychiatric drugs. Titled “Initiation and Adaptation: A Paradigm for Understanding Psychotropic Drug Action,” it was published in the American Journal of Psychiatry, and it told of how all psychotropic drugs could be understood to act on the brain in a common way.46
Antipsychotics, antidepressants, and other psychotropic drugs, he wrote, “create perturbations in neurotansmitter functions.” In response, the brain goes through a series of compensatory adaptations. If a drug blocks a neurotransmitter (as an antipsychotic does), the presynaptic neurons spring into hyper gear and release more of it, and the postsynaptic neurons increase the density of their receptors for that chemical messenger. Conversely, if a drug increases the synaptic levels of a neurotransmitter (as an antidepressant does), it provokes the opposite response: The presynaptic neurons decrease their firing rates and the postsynaptic neurons decrease the density of their receptors for the neurotransmitter. In each instance, the brain is trying to nullify the drug’s effects. “These adaptations,” Hyman explained, “are rooted in homeostatic mechanisms that exist, presumably, to permit cells to maintain their equilibrium in the face of alterations in the environment or changes in the internal milieu.”
However, after a period of time, these compensatory mechanisms break down. The “chronic administration” of the drug then causes “substantial and long-lasting alterations in neural function,” Hyman wrote. As part of this long-term adaptation process, there are changes in intracellular signaling pathways and gene expression. After a few weeks, he concluded, the person’s brain is functioning in a manner that is “qualitatively as well as quantitatively different from the normal state.”
His was an elegant paper, and it summed up what had been learned from decades of impressive scientific work. Forty years earlier, when Thorazine and the other first-generation psychiatric drugs were discovered, scientists had little understanding of how neurons communicated with one another. Now they had a remarkably detailed understanding of neurotransmitter systems in the brain and of how drugs acted on them. And what science had revealed was this: Prior to treatment, patients diagnosed with schizophrenia, depression, and other psychiatric disorders do not suffer from any known “chemical imbalance.” However, once a person is put on a psychiatric medication, which, in one manner or another, throws a wrench into the usual mechanics of a neuronal pathway, his or her brain begins to function, as Hyman observed, abnormally.
Back to the Beginning
While Dr. Hyman’s paper may seem startling, it serves as a coda to a scientific narrative that is, in fact, consistent from beginning to end. His was not a conclusion that should be seen as unexpected, but rather one that was predicted by psychopharmacology’s opening chapter.
As we saw, Thorazine, Miltown, and Marsilid were all derived from compounds that had been developed for other purposes—for use in surgery or as possible “magic bullets” against infectious diseases. Those compounds were then found to cause alterations in mood, behavior, and thinking that were seen as helpful to psychiatric patients. The drugs, in essence, were perceived as having beneficial side effects. They perturbed normal function, and that understanding was reflected in the initial names given to them. Chlorpromazine was a “major tranquilizer,” and it was said to produce a change in being similar to frontal lobotomy. Meprobamate was a “minor tranquilizer,” and in animal studies, it had been shown to be a powerful muscle relaxant that blocked normal emotional response to environmental stressors. Iproniazid was a “psychic stimulator,” and if the report of TB patients dancing in the wards was truthful, it was a drug that could provoke something akin to mania. However, psychiatry then reconceived the drugs as “magic bullets” for mental disorders, the drugs hypothesized to be antidotes to chemical imbalances in the brain. But that theory, which arose as much from wishful thinking as from science, was investigated and it did not pan out. Instead, as Hyman wrote, psychotropics are drugs that perturb the normal functioning of neuronal pathways in the brain. Psychiatry’s first impression of its new drugs turned out to be the scientifically accurate one.
With this understanding of psychiatric medications now in mind, it is possible to pose the scientific question at the heart of this book: Do these drugs help or harm patients over the long term? What do fifty years of outcomes research show?
* The NIMH researchers also looked at a number of other possible associations between variable neurotransmitter levels and response to an antidepressant. They measured norepinephrine metabolites and dopamine metabolites; they divided their depressed patients into bipolar and unipolar groups; and they evaluated their response to two antidepressants, imipramine and amitriptyline. They found mild associations between several of these subgroups and their response to one or other of the drugs; I have focused here on their findings regarding whether (a) depression is due to low levels of serotonin, and (b) if the subgroup of patients with low levels of serotonin responds better to a drug that selectively blocks the reuptake of this neurotransmitter.
* Over the long term, it appears that serotonin release falls to an abnormally low level, at least in certain regions of the brain.