5
Cholesterol (C27H46O) is a waxy organic fat-soluble substance (or lipid), which is an essential structural component of the membranes of all animal (but not plant) cells (Figure 5.11).
The cholesterol molecule.
It is synthesized mainly in the liver and is abundant in diets containing meat, dairy, and other animal products. In addition to its structural role, cholesterol is also the common precursor of bile acids and all the steroid hormones, including cortisone and the sex hormones, which are synthesized in the adrenal glands, testes, and ovaries.
Because it is not soluble in water, cholesterol requires “carrier” proteins (apolipoproteins A, B, and others) to emulsify it so that it can travel through the blood in particles called lipoproteins. Lipoproteins were discovered in the 1920s and classified by their density, which is assessed by the degree to which they sink or float in an ultracentrifuge. In general, lipoproteins with high protein and low fat content sink and those with low protein and high fat content float. There are four broad classes of lipoproteins (going from heaviest to lightest): (a) high-density lipoprotein (HDL), which contains mainly apo-A and cholesterol, (b) low-density lipoprotein (LDL), which contains mainly apo-B and cholesterol, (c) very low-density lipoprotein (VLDL), which contains apo-B, C, and E and cholesterol and large amounts of triglyceride, and (d) triglyceride-rich chylomicrons, which transport dietary fats from the small intestine and are normally found in the blood only after meals. In the typical American or European, at least 65% of the circulating cholesterol is carried by LDL. In 1967, Fredrickson et al. classified the pathologic states associated with high lipoprotein levels.1 Their second category (type II), in which LDL levels are elevated, will concern us most in this chapter.
Felix Marchand coined the term atherosclerosis in 1904, to describe the inflammatory changes that had been observed in the blood vessel walls of patients dying of cardiovascular causes fifty years earlier by the pathologists Karl von Rokitansky and Rudolf Virchow.2 The term refers to the hardness of the artery (“sclerosis”) and to the gruel-like consistency (“athero”) of the substance (cholesterol, fats, and cellular debris) extruded when the lesion (or plaque) is cut. Figure 5.2 depicts an atherosclerotic plaque that partially obstructs an artery longitudinally and in cross section. From there, it was an easy leap to tie clinical cardiovascular events like heart attack and stroke, which involve the sudden and catastrophic obstruction of the blood supply. of vital organs like the heart and brain, to atherosclerosis.
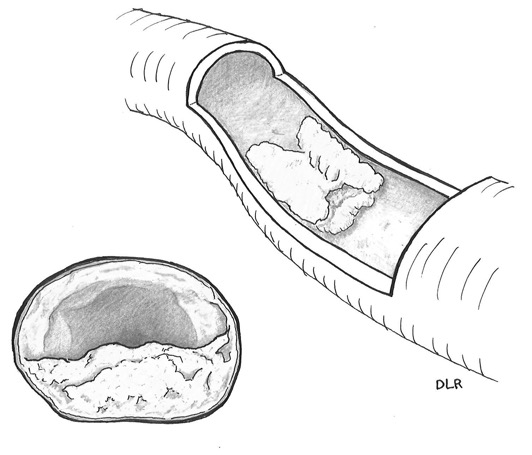
Longitudinal and cross-sectional drawings of an atherosclerotic plaque. Artwork by Debra L. Roney.
The “cholesterol hypothesis” placing cholesterol in the causal chain for atherosclerotic cardiovascular disease got its start in 1913, when the Russian pathologist Nikolai Anichkov was able to reproduce atherosclerosis in rabbits by feeding them cholesterol that had been extracted and purified from egg yolks.3 This is not easy to replicate experimentally in humans, in whom you can look at circulating cholesterol levels, but not—at least not easily—the lesions themselves. In the classical human feeding study by Ancel Keys, the short-term impact of dietary cholesterol intake on serum cholesterol was modest; the relative amounts of dietary saturated fats (hard fats typically found in meats, butter, and tropical plants) and unsaturated fats (soft fats and oils typically found in plants and fish) had a larger impact.4 Other circumstantial evidence supported the cholesterol hypothesis:
1. The transitory 30% decline in cardiovascular mortality rates in Norway and other European countries occupied by Germany during World War II, when meat, dairy products, and eggs were in short supply.5
2. The Honolulu Heart Study of Japanese migrants, which showed a rise heart attack rates and fall in stroke rates as migrants adopted a western diet and lifestyle.6
3. The Seven Countries Study, which compared the populations of the U.S., Canada, Australia, England, Wales, Italy and Japan and found higher cardiovascular mortality rates in the countries consuming the most animal fat.7
However, none of this evidence came close to being dispositive. By the time Framingham and other prospective epidemiologic studies identified serum cholesterol as a major cardiovascular risk factor in the 1960s, medical science had little to offer for the treatment and control of high blood cholesterol and could offer no definitive proof that doing so would prevent heart attacks and other cardiovascular events. Until the development of the statins in the late 1980s the only approved cholesterol-lowering drugs were:
1. The bile acid sequestrant resins cholestyramine and colestipol, which inhibit the reabsorption of bile acids (a product of cholesterol) in the small intestine, thus forcing the liver to divert some cholesterol to the production of more bile acids. These drugs can theoretically lower LDL cholesterol by 25%, but they were administered as a gritty slurry (earning them the nickname “sand”), which must be drunk in large daily quantities and often cause constipation and abdominal discomfort. The resins remain within the intestine and do not enter the body and are therefore quite safe; nevertheless, patient compliance is problematic.
2. Niacin, a vitamin which moderately (10%) lowers LDL and VLDL and raises HDL cholesterol, when given in high doses. Niacin also has significant vasomotor side effects (hot flushes), which limit patient compliance.
3. Clofibrate, which has a similar pharmacological effect to high-dose niacin but fewer side effects. Two newer fibrate drugs, gemfibrozil and fenofibrate, are in use today.
Until the mid–1970s, most of the trials using these drugs and/or cholesterol-lowering diets were secondary prevention trials in a few hundred patients. Blood cholesterol was typically lowered by 10% or less, and the results were mixed.8 The largest trial was the Coronary Drug Project initiated by the NIH in 1966.9 This double-blind placebo-controlled randomized trial in 8341 men with known heart disease had five active treatment arms (a) niacin, (b) clofibrate, (c) dextrothyroxine (a thyroid hormone analog), (d) low-dose estrogen, and (e) high-dose estrogen. None of the five drugs reduced mortality (the primary outcome). The men treated with niacin did show a significant favorable trend in recurrent coronary events.10 But the dextrothyroxine and estrogen treatment arms had to be stopped early due to net adverse trends in cardiovascular outcomes.11 (It should be noted that estrogen was used in this trial to mimic the relative “protection” of women from atherosclerotic cardiovascular disease, not to lower cholesterol levels.)
The World Health Organization (WHO) also initiated a large (15,763 participants) primary prevention trial using clofibrate in Edinburgh, Prague, and Budapest in 1962.12 After five years of treatment, 25% more deaths were observed in the clofibrate than in the placebo group. However, after 13.5 years of extended follow up, mortality did not differ between groups.13 Although the dismal results of the CDP and WHO clofibrate trials removed clofibrate as a viable cholesterol-lowering drug, two newer fibrate drugs, gemfibrozil and fenofibrate, have fared better in subsequent trials.14
The Lipid Research Clinics Coronary Primary Prevention Trial (LRC-CPPT)
In 1973, the NHLBI, after rejecting the concept of a large diet-heart trial as unfeasible, initiated the Lipid Research Clinics Coronary Primary Prevention Trial (LRC-CPPT), a double-blind placebo-controlled primary prevention trial of cholestyramine in 3806 35- to 59-year-old men with type 2 hypercholesterolemia (LDL cholesterol > 175 mg/dL after three months of dietary treatment).15 The study, which cost more than $150 million in circa 1980 dollars, was among the most expensive cardiovascular trials ever done. As a junior member of the NHLBI’s LRC-CPPT management team, I can personally attest that no effort or expense were spared in the execution of this trial. Not one of the 3806 trial participants were lost to follow-up, and all cardiovascular hospitalizations and deaths were independently reviewed by a blinded panel of cardiologists to insure rigorous and objective classification of study endpoints. The trial had the strong backing of Donald Fredrickson and Bob Levy, two of the architects of the lipoprotein classification system, who had risen to the directorship of the NIH and NHLBI, respectively. Many of the twelve Lipid Research Clinics at which the trial was performed were alumni of Fredrickson’s NIH laboratory, who had become eminent researchers in their own right. The data coordinating center was set up at the University of North Carolina, one of the country’s leading schools of public health, and the Centers for Disease Control (CDC) standardized all cholesterol and lipoprotein measurements. The Lipid Metabolism Branch of the NHLBI, which was led by Dr. Basil Rifkind after Dr. Levy assumed the NHLBI directorship, was abundantly staffed with clinical trials experts, epidemiologists, nutritionists, and laboratory scientists who helped manage the trial.
However, when the much-anticipated trial results were announced in January 1984, they were underwhelming.16 The difference in the primary outcome, the combined incidence of fatal and nonfatal heart attacks, was small—155 in the cholestyramine group versus 187 in the placebo group—and of marginal statistical significance. Although this 17% relative reduction in the primary endpoint was supported by similar reductions in secondary endpoints like stroke, other adverse cardiovascular outcomes, and the conversion of annual graded exercise tests from negative to positive, there was essentially no difference in total mortality—68 deaths in the cholestyramine group versus 71 deaths in the placebo group. The main problem was that cholestyramine, which lowered cholesterol by as much as 25% in small short-term trials, produced only a mean 8% cholesterol reduction in the LRC-CPPT due to deteriorating compliance over the trial’s 7–10-year duration. The investigators pointed out that a 17% reduction in cardiac events was almost exactly what predictive models (like Framingham) would have anticipated for an 8% cholesterol reduction. A model based on internal analysis of the trial data showed o 49% reduction in heart attacks among LRC-CPPT participants who did maintain a 25% cholesterol reduction.17 However, the LRC-CPPT certainly did not offer the clarity of the early hypertension trials and did not boost prescriptions of cholestyramine or other available cholesterol-lowering drugs. The cholesterol hypothesis seemed to be stuck and might have remained so but for two developments in the laboratory while the LRC-CPPT was underway—the identification and elucidation of the LDL receptor gene, for which Michael Brown and Joseph Goldstein won the 1985 Nobel Prize in Medicine, and the discovery in Japan of an antibiotic secreted by a penicillium fungus, which eventually became the prototype statin drug.
Familial Hypercholesterolemia and the LDL Receptor Gene
A Norwegian clinician, Carl Muller, first recognized familial hypercholesterolemia (FH) as an autosomal dominant inherited disorder characterized by high serum cholesterol and early severe cardiovascular disease in 1938.18 Children who inherit copies of the mutated gene from both parents (homozygotes) have serum cholesterol levels approaching 1000 mg/dL, resulting in cholesterol deposits in their skin, tendons, and corneas, and severe atherosclerotic cardiovascular disease, manifesting in heart attacks and often death before they reach adulthood. Since these severely affected children respond poorly to cholesterol-lowering drugs, their treatment relies on plasma apheresis, a dialysis-like procedure in which the non-cellular (plasma) portion of the blood is cleansed of LDL perhaps twice a week. Persons who inherit a mutated gene from one parent and a normal gene from the other have milder manifestations with cholesterol levels in the 300–400 mg/dL range; these persons do respond to cholesterol-lowering drugs but still tend to have heart attacks in middle age and to die prematurely. The combined frequency of the many FH mutation variants is about one in 1000 in the U.S. This means that the prevalence of the severe homozygous form of the disease is about one in a million (the probability of inheriting a defective gene from both parents), while the milder heterozygotes comprise about 0.2% of the population, or more than 600,000 Americans. Brown and Goldstein demonstrated that the mutations that cause FH entail alterations in the structure of receptor molecules that enable LDL to move from the blood into the liver and other organs where the cholesterol it contains can be repurposed for the production of hormones and other essential molecules.19 If these receptors are disabled, the liver and other organs must make their own cholesterol, while LDL levels in the bloodstream climb. According to the cholesterol hypothesis, these high circulating levels of LDL promote atherosclerosis. Thus, upregulating the production of LDL receptors in patients who have them would be an effective therapeutic strategy to reduce circulating LDL levels and inhibit atherosclerosis.
The Statins
In April 1971, Akira Endo, a Japanese pharmacologist employed by Sankyo, became interested in finding an antibiotic that would inhibit the synthesis of cholesterol and, inspired by the legendary discovery of penicillin by Alexander Fleming, began screening 3800 species of fungi for suitable compounds.20 After about a year, his research produced citrinin, a potent inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase, the key rate-controlling enzyme in the synthetic pathway for cholesterol. Although animal experiments showing severe kidney toxicity quickly ruled out the use citrinin in humans, its discovery paved the way for the discovery of a second active compound called compactin several months later. For reasons never fully explicated, Sankyo abandoned compactin in 1980 after several years of animal studies and sold the rights to Merck, who developed a similar compound of its own called mevinolin (or lovastatin) with a superior safety profile. After years of testing, the U.S. FDA approved the clinical use of lovastatin for lowering LDL cholesterol in 1987, although randomized clinical trials were still needed to prove its efficacy in reducing rates of heart attack and other cardiovascular events. Still, because of its relative freedom from side effects and its ability to lower serum cholesterol by 20–30% under real-world conditions, prescriptions of lovastatin quickly surpassed the resins. Lovastatin was soon joined and surpassed by many other statin drugs—pravastatin (Pravachol), simvastatin (Zocor), fluvastatin (Leschol), atorvastatin (Lipitor), rosuvastatin (Crestor), pitavastatin (Livalo)—which offered advantages in potency and/or side effect profiles. According to ClinCalc, atorvastatin, simvastatin, pravastatin, and rosuvastatin were the second, eighth, 26th, and 39th most prescribed drugs in the U.S. in 2017, with more than 205 million prescriptions among them.21
The case for the cholesterol hypothesis was further bolstered in 1990 by the report of an unusual randomized trial in which partial ileal bypass surgery, in which the final segment of the small intestine (where bile acids are reabsorbed) is bypassed by joining the previous segment of the small intestine directly to the colon.22 This procedure is tantamount to a more drastic and permanent version of cholestyramine resin. As one might imagine, recruitment was difficult and protracted; only 838 patients were randomized over nearly eight years. However, because the surgery produced a sustained 24% cholesterol reduction, there was a highly significant 35% reduction in the combined incidence of fatal and nonfatal MI, although the reductions in the two primary outcomes—mortality from coronary disease and from all causes—fell short of statistical significance. While the results of this trial provided proof of concept that a treatment that could actually produce a sustained cholesterol reduction of 20% or more could prevent heart attacks, the actual treatment used in this trial had little practical value.
So, returning to the mid–1980s, the NHLBI could have aptly portrayed LRC-CPPT results as hopeful but not definitive while awaiting results from the ongoing statin trials to perhaps provide the required proof. But they chose a riskier and more controversial route, promoting the LRC-CPPT results as having definitively provided the last missing piece to prove the cholesterol hypothesis. The NHLBI leadership argued that the LRC-CPPT findings should be viewed in the broader context of evidence from epidemiology and the laboratory (including the findings of Brown and Goldstein), and pointed to the development of new and more powerful cholesterol-lowering drugs (the statins) looming on the horizon. They established the National Cholesterol Education Program to promote public awareness of cholesterol and issued treatment guidelines (analogous to those for BP) which mostly emphasized diet and then stepped back to await the results of the pharmaceutical industry’s ongoing statin trials.23 While the NHLBI’s promotional efforts laid the groundwork for the statins, they stirred up a firestorm of controversy.24 Some of the objections were arcane (e.g., quibbles over the criteria for statistical significance), and others were silly (e.g., attributing biological importance to the flukish 11–4 excess of deaths by accident, suicide, or homicide in the cholestyramine group). However, critics of the LRC-CPPT included many serious scientists who were justifiably skeptical of orchestrating an entire public health campaign based on the trial of a treatment that few patients would tolerate for a week (let alone a lifetime), which showed only a small, marginally significant benefit at best, and which was then extrapolated to cholesterol-lowering therapies that had not even been tested yet. This approach could easily have backfired if the statin drugs had not panned out.
But the statin drugs did pan out—and in a big way. In November 1993, Merck announced the results of its Scandinavian Simvastatin Survival Study (SSSS) in 4444 patients with prior heart attacks, in which simvastatin significantly reduced heart attacks, heart attack deaths, and total mortality by 34%, 42%, and 30% respectively.25 A year later, Parke Davis announced similarly positive results of their West of Scotland Coronary Prevention Study (WOSCOPS) of pravastatin in 6595 hypercholesterolemic men without prior heart disease.26 Twelve additional statin trials with acronyms like CARE (pravastatin) Post-CABG (lovastatin in patients with coronary bypass grafts), AFCAPS/TEXCAPS (lovastatin) LIPID (pravastatin), GISSI-P (pravastatin, not blinded), LIPS (fluvastatin), HPS (simvastatin), PROSPER (pravastatin), ALLHAT-LLT (pravastatin, not blinded), ASCOT-LLA (atorvastatin), ALERT (fluvastatin),and CARDS (atorvastatin) all reported results in the ensuing decade, and were included in the 2005 report of the Oxford Cholesterol Treatment Trialists prospective meta-analysis, which included 14 statin trials with 90,056 participants.27 Except for the two non-blinded trials, where there were high rates of crossover to statin treatment in the control group, all reported statistically significant reductions in major vascular events. When the results were expressed in terms of risk reduction per 1.0 mMol/dL (38.6 mg/dL) lowering of LDL cholesterol, the results fit a straight line with a slope of 23% reduction in risk of major coronary events (mainly heart attacks) per mmol/dL decrease in LDL cholesterol. Similar reductions were observed for other cardiovascular events, including strokes and heart attack deaths. Statins had no adverse effect on cancer or other causes of death; mortality from all causes was significantly reduced by 12%. The percent reduction in major vascular diseases was similar and robust across patient subgroups—for example, 22% in patients with a prior heart attack versus 28% in patients with no prior heart disease, 26% in patients below age 65 versus 19% in seniors, 24% in men versus 18% in women, 21% in hypertensive patients versus 25% in others, 22% in diabetics versus 23% in others, etc.
In an updated 2010 analysis of 170,000 participants, which included twelve additional statin trials (five of which compared high- versus low-dose statins), the Cholesterol Treatment Trialists found more of the same.28 In the 21 placebo-controlled trials, a mean 1.07 mmol/dL (41.3 mg/dL) mean reduction in LDL cholesterol, brought a highly significant 22% decrease in major vascular events (heart attacks, strokes, etc.). The results of these 21 trials are plotted against the LDL cholesterol reduction after one year is provided in Figure 5.3.
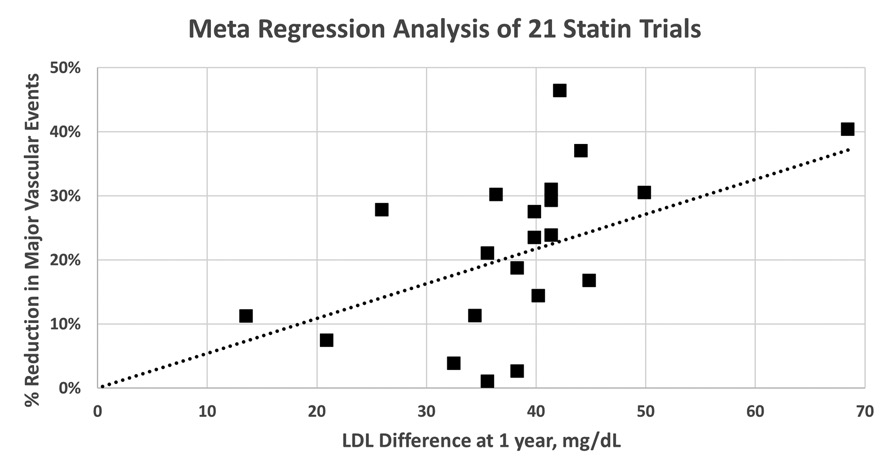
Meta-regression analysis of 21 statin trials. Percent reduction in risk of major vascular events for 21 statin trials is plotted versus LAD cholesterol difference at one year. The trials are SSSS, WOSCOPS, CARE, Post-CABG, AFCAPS/TexCAPS, LIPID, GISSI-P, LIPS, HPS, PROSPER, ALLHAT-LLT, ASCOT-LLA, ALERT, CARDS, ALLIANCE, 4D, ASPEN, MEGA, JUPITER, GISSI-HF, and AURORA. Data were obtained from the Cholesterol Treatment Trialists (CTT) Collaboration, Lancet 2010; 376: 1670–1681. The dashed line represents a weighted linear regression in which each trial was weighted in inverse proportion to the standard error of its estimated risk reduction.
Although there is considerable variability around the regression line, the general trend comes through—the greater the reduction in LDL cholesterol, the greater the reduction in risk. In the five trials of intensive (high-dose and or high-potency) statin treatment, an additional 0.51 mmol/dL (20 mg/dL) mean reduction in LDL cholesterol brought an additional significant 15% reduction in major vascular events. In general, reductions of 22% in major vascular events and 10% in total mortality were observed for every 1 mmol/L (38.6 mg/dL) reduction in LDL cholesterol. Thus, a patient receiving intensive statin therapy who attained the typical 1.58 mmol/L (61 mg/dL) decrease in LDL cholesterol (versus placebo) can expect to attain a net 32% reduction in the risk of major vascular events and a 15% reduction in risk of death. The risk reductions were again similar in all relevant subgroups—men and women, older and younger persons, persons with and without prior cardiovascular disease, persons with or without diabetes or hypertension, and persons starting from different baseline LDL cholesterol levels. There was no increase in cancer or other non-cardiovascular deaths in participants who received statins. Although statins still have their detractors and no drug is tolerated by everyone, no class of drug has been studied more thoroughly and rigorously than the statins regarding safety and efficacy in as many persons and has emerged with as clean a slate. The worst that can be said for the statins is that they can cause significant muscle inflammation (which can lead to kidney failure and death in extremely rare cases) in a small fraction of persons. Also, statins cause small increases in blood sugar that may complicate their use in diabetics; however, statins are the single best class of drugs for preventing major vascular events—the leading cause of death—in diabetics.29
Several non-statin cholesterol-lowering drugs have also emerged in the 21st century. Ezetimibe (Zetia), an oral agent approved in 2002, is often used in conjunction with a statin to add another 10–15% reduction in LDL cholesterol level. The most promising new drugs are the proprotein convertase subtilisin kexin 9 (PCSK9) blockers evolocumab (Repatha) and alirocumab (Praluent), which are monoclonal immunoglobulins that help the liver clear LDL cholesterol by blocking a protein called PCSK9, which interferes with LDL receptors.30 These drugs, which can be given only by injection, mimic a naturally occurring human PCSK9 mutation which is characterized by very low levels of LDL cholesterol and correspondingly low rates of cardiovascular disease. PCSK9 blockers are especially useful in FH heterozygotes whose LDL cholesterol levels remain elevated even after taking statins; when they are used in combination with a statin, LDL cholesterol reductions of 65% or more may be achieved. However, since the efficacy of all these drugs requires LDL receptors, they are not very effective in FH homozygotes. The recent approval of lomitapide (Juxtapid), which partially blocks the production of LDL particles, may offer these patients some relief.
In summary, the treatment of high BP and high LDL cholesterol are the twin bulwarks of modern preventive cardiology. The BP and cholesterol stories have many parallels but also some important differences. The major randomized trials establishing the importance of BP lowering were completed by 1992, before the first major statin trial was published, while people were still arguing over whether the cholesterol hypothesis was a myth. While physicians had their choice of multiple classes of effective BP drugs that were generally used in combinations, physicians had no good options for lowering LDL cholesterol until the statins came along and even today rely primarily on the statins. The NHLBI has remained a central player in BP trials into the 21st century (SPRINT) but has played only a minor supporting role in cholesterol trials since 1984.
Impact of Cholesterol-Lowering on Heart Attack Deaths
Like hypertension, the treatment of high blood cholesterol has traveled a long road from acrimonious controversy to consensus, but within half the time. Unlike hypertension, the road was for cholesterol was tortuous and laden with potholes. As recently as 1990, the state of cholesterol lowering drugs was little better than that of antihypertensive drugs in 1960, and some dismissed the cholesterol hypothesis as myth. It was not until 1994 that the first clinical trial demonstrated that statin treatment could prevent cardiovascular events, and it was not until 2001 that the National Cholesterol Education Program Expert Panel was able to give a full-throated endorsement to statins as the first line of pharmacologic treatment of high blood cholesterol.31 In 1988–94, just before the first statin trial was published, only 3.4% of U.S. adults reported taking a cholesterol-lowering drug of any kind; that percentage grew to 9.3% in 1999–2002 and 15.5% in 2007–2010.32 The percentage of American adults using statins doubled between 1999–2002 and 2010–11, growing from 7.4% to 15.9% in men and from 9.6% to 18.9% in women.33 If one just looks at Americans over age 40, the prevalence of cholesterol-lowering drugs grew from 19.9% in 2003–04 to 27.9% in 2011–12, while the prevalence of statin use grew from 16.3% to 23.2% over the same decade.34
In 2018, the ACC/AHA Guidelines for cholesterol treatment have established LDL cholesterol < 100 mg/dL as the ideal and recommend intensive statin treatment to achieve that goal for patients without cardiovascular disease whose 10-year cardiovascular risk exceeds 7.5%.35 They further recommend intensive statin treatment for all patients with established cardiovascular disease, with a goal of reducing LDL cholesterol by 50%. A recent analysis of 2012–13 data showed that while statin use was 27.9% among all Americans over age 40, roughly 60% of those with high risk due to existing cardiovascular disease and 50% of those with diabetes were using a statin.36 The use of statins in secondary prevention has grown from zero in 1980 to 45% in 2000 to 72% in 2011.37
The IMPACT model of Ford et al. attributes 24.2% of the observed decline in heart attack mortality between 1980 and 2000—to the reduction in the mean population LDL cholesterol level in the U.S. population from 5.67 mmol/L in 1980 to 5.33 mmol/L in 2000, which according to the Cholesterol Treatment Trialists meta-analysis would translate to a 32% reduction in heart attack deaths in men and a 31% reduction in women.38 Most of this reduction was likely due to the introduction of statins in the late 1980s. The model also estimated that statin treatment in various secondary prevention settings (after a heart attack, heart failure, coronary bypass surgery, hypertension, etc.) accounted for an additional 8.5% of the observed reduction in heart attack mortality. Thus, the introduction of statins drugs accounted for roughly one-third of the 46% reduction in heart attack deaths (from 345.2 to 186.8 deaths per 100,000) between 1980 and 2000. But what about the contribution of statins to the accelerating decline in heart attack mortality after 2000, as shown in Figure 1.2?
I have applied the method described for hypertension in Chapter 4 (and explained in detail in the appendix) to extrapolate the published IMPACT findings for statins to cover the 77.4% decline in heart attack mortality in 1968–2011 (the most recent year for which I have national statin usage data for all adults). Based on the increase in statin usage by a factor of 2.05 between 2000 and 2011, I estimate that LDL cholesterol lowering—mainly statin usage—in the general population (i.e., primary prevention) has accounted for 27.7% of the 77.4% decline in heart attack deaths between 1968 and 2011 (see Table A.2). Similarly, I estimate the increase in statin usage in persons who have already had a heart attack (i.e., secondary prevention) by a factor of 1.71 between 2000 and 2011, has accounted for 8.2% of this mortality decline (see Table A.4). All told, 35.9% of the 1968–2011 decline in heart attack mortality can be attributed to statins and other cholesterol lowering interventions. These figures probably understate the total cardiovascular impact of statins since they do not include reduction of atherothrombotic strokes. (Cholesterol lowering does not reduce strokes caused by intracranial bleeding.) It also does not model the increasing use of more aggressive statin regimens in secondary prevention and high-risk primary prevention, as recommended in the newest cholesterol treatment guidelines. All in all, it is fair to state that the development and promulgation of statins during the past three decades is probably the single greatest factor in the decline in heart attack deaths since 1968.
1. DS Fredrickson, RI Levy, RS Lees. Fat Transport in Lipoproteins—An integrated approach to mechanisms and disorders. N Engl J Med 1967; 276:273–281, DOI: .1056/NEJM196702022760507.
2. AM Gotto, Jeremiah Metzger Lecture: Cholesterol, Inflammation and Atherosclerotic Cardiovascular Disease: Is it all LDL? JL Goldstein, MS Brown. A century of cholesterol and coronaries. From plaques to genes to statins. Cell 2015; 161:161–172. doi: 10.1016/j.cell.2015.01.036.
3. NN Anichkov, S Chalatow. Ueber experimentelle Cholesterinsteatose und ihre Bedeuting fur die Entstehung einer pathologischer Prozesse. Zentralbl Allg Pathol 1913; 24:1–9.
4. A Keys, JT Anderson, F Grande. Serum cholesterol response to changes in the diet. Metabolism 1965; 14:747–787.
5. University of Minnesota Driven to Discover. Heart Attack Prevention: A History of Cardiovascular Epidemiology—Mortality Statistics. http://www.epi.umn.edu/cvdepi/history-gallery/mortality-statistics/.
6. RM Worth, H Kato, GG Rhoads, A Kagan, SL Syme. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: mortality. Am J Epidemiol 1975; 102:481–490.
7. A Keys, C Aravanis, H Blackburn, R Buzina, BS Djordjevic, AS Dontas, F Fidanza, MJ Karvonen, N Kimura, A Menotti , I Mohacek, S Nedeljkovic, V Puddu, S Punsar, HL Taylor, FSP Van Buchem. Seven countries. A Multivariate Analysis of Death and Coronary Heart Disease. Cambridge: Harvard University Press, 1980.
8. University of Minnesota Driven to Discover. Heart Attack Prevention: A History of Cardiovascular Epidemiology—Study Synopses. October 15, 2012. http://www.epi.umn.edu/cvdepi/the-research/study-synopses/?search_keyword=&search_title=yes&search_synopsis=yes&search_people=&search_study_topic=&search_study_category=527&filter_search=yes#results-title.
9. ClinicalTrials.gov. Coronary Drug Project. https://clinicaltrials.gov/ct2/show/NCT00000482?term=coronary+drug+project&draw=2&rank=2.
10. The Coronary Drug Project. Clofibrate and niacin in coronary heart disease. JAMA 1975, Jan 27; 231(4):360–81.
11. The Coronary Drug Project. Initial findings leading to modifications of its research protocol. JAMA 1970, Nov 16; 214(7):1303–1313.
The Coronary Drug Project. Findings leading to further modifications of its protocol with respect to dextrothyroxine. The coronary drug project research group. JAMA 1972, May 15; 220(7):996–1008.
12. M Oliver. The clofibrate saga: a retrospective commentary. Br J Clin Pharmacol 2012; 74:907–910. doi: 10.1111/j.1365–2125.2012.04282.x.
13. WHO cooperative trial on primary prevention of ischaemic heart disease with clofibrate to lower serum cholesterol: final mortality follow-up. Lancet 1984; 2(8403):600–604.
14. MH Frick, O Elo, K Haapa, OP Heinonen, P Heinsalmi, P Helo, JK Huttunen, P Kaitaniemi, P Koskinen, V Manninen, H Maenpaa, M Malkonen, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 1987; 317:1237–1245. HB Rubins, SJ Robins, D Collins, CL Fye, JW Anderson, MB Elam, FH Faas, E Linares, EJ Schaefer, G Schectman, TJ Wilt, J Wittes. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 1999; 341:410–418. The ACCORD Study Group. The effect of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563–1574. DOI: 10.1056/NEJMoa1001282.
15. ClinicalTrials.gov. Lipid Research Clinics Coronary Primary Prevention Trial (LRC-CPPT). https://clinicaltrials.gov/ct2/show/NCT00000488?term=Lipid+Research+Clinics&draw=2&rank=2.
16. The Lipid Research Clinics Program. The Lipid Research Clinics Coronary Primary Prevention Trial Results I. Reduction in incidence of coronary heart disease. JAMA 251: 351–364, 1984.
17. The Lipid Research Clinics Program. The Lipid Research Clinics Coronary Primary Prevention Trial Results II. The relationship of reduction in incidence of coronary heart disease to cholesterol reduction. JAMA 1984; 251 365–374.
18. JL Goldstein, MS Brown. History of Discovery: The LDL Receptor. Arterioscler Thromb Vasc Biol 2009; 29:431–438.
19. MS Brown, JL Goldstein. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
20. MS Brown, JL Goldstein JL. A tribute to Akira Endo, discoverer of a “Penicillin” for cholesterol. Atherosclerosis Suppl 2004; 5:13–6. A Endo. A historical perspective on the study of statins. Proc Jpn Acad Ser B 2010; 86:484–493.
21. ClinCalc DrugStats Database. The top 200 drugs of 2020. https://clincalc.com/DrugStats/Top200Drugs.aspx.
22. H Buchwald, RL Varco, JP Matts, JM Long, LL Fitch, GS Campbell, MB Pearce, AE Yellin, WA Edmiston, RD Smink, HS Sawin, CT Campos, BJ Hansen, N Tuna, JN Karnegis, ME Sanmarco, K Amplatz, WR Casteneda-Zuniga, DW Hunter, JK Bissett, FW Weber, JW Stevenson, AS Leon, TC Chalmers, and the POSCH Group. Effect of Partial Ileal Bypass Surgery on Mortality and Morbidity from Coronary Heart Disease in Patients with Hypercholesterolemia—Report of the Program on the Surgical Control of the Hyperlipidemias (POSCH). N Engl J Med 1990; 323:946–355, DOI: 10.1056/NEJM199010043231404.
23. Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. The Expert Panel. Arch Intern Med 1988; 148:36–69.
24. TJ More. The Cholesterol Myth. Atlantic Monthly, September 1989, pp. 37–70.
25. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
26. J Shepherd, SM Cobbe, I Ford, CG Isles, AR Lorimer, PW MacFarlane, JH McKillop, CJ Packard, West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med 1995; 333:1301–07.
27. Cholesterol Treatment Trialists Collaboration. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet 2005; 366:1267–1278.DOI:10.1016/S0140–6736(05)67394–1.
28. Cholesterol Treatment Trialists Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
29. Cholesterol Treatment Trialists Collaboration. Efficacy of cholesterol-lowering therapy in 18 686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
30. AS Peterson, LG Fong, SG Young. PCSK9 function and physiology. J Lipid Res 2008; 49:1152–1156. doi: 10.1194/jlr.E800008-JLR200.
31. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Executive Summary. National Cholesterol Education Program, National Heart, Lung, and Blood Institute, National Institutes of Health. NIH Publication No. 01–3670, May 2001.
32. MD Carroll, AK Kit, DA Lacher, ST Shero, ME Mussolino. Trends in lipids and lipoproteins of U.S. adults, 1988–2010. JAMA 2012; 308:1545–1554.
33. EM Sarpong, SH Zuvekas. Changes in statin therapy among adults (age 18+) by selected characteristics, United States, 2000–2001 to 2010–2011. Medical Expenditure Panel Survey (MEPS). Statistical Brief #459. November 2014. https://www.ncbi.nlm.nih.gov/books/NBK470833/.
34. Q Gu, R Paulose-Ram, VL Burt, BK Kit. Prescription cholesterol-lowering medication use in adults aged 40 and over: United States, 2003–2012. NCHS Data Brief No. 177, December 2014. https://www.cdc.gov/nchs/data/databriefs/db177.pdf.
35. SM Grundy, NJ Stone, AL Bailey, C Beam, KK Birtcher, RS Blumenthal, LT Braun, S de Ferranti, J Faiella-Tommasino, DE Forman, R Goldberg, PA Heidenreich, MA Hlatky, DW Jones, D Lloyd-Jones, N Lopez-Pajares, CE Ndumele, CE Orringer, CA Peralta, JJ Saseen, SC Smith, L Sperling, SS Virani, J Yeboah. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019; 139:e1082-e1143. https://www.ahajournals.org/doi/10.1161/CIR.0000000000000625.
36. JA Salami, H Warralch, J Velardo-Elizondo, ES Spatz, NR Desai, JS Rana, SS Virani, R Blankstein, A Khera, MJ Blaha, RS Blumenthal, D Lloyd-Jones, K Nasir. National trends in statin use and expenditures, in the U.S. adult population from 2002 to 2013: Insights from the Medical Expenditure Panel survey. JAMA Cardiology 2017; 2:56–65. doi.10.1001/jamacardio.2016.4700. file:///C:/Users/gordo/AppData/Local/Temp/jamacardiology_salami_2016_oi_160082.pdf.
37. NS Shah, MD Huffman, H Ning, DM Lloyd-Jones. Trends in myocardial infarction secondary prevention: The National Health and Nutrition Examination Surveys (NHANES), 1999–2012. J Am Heart Assoc 2015; 4:1–12. doi:10.1161/JAHA.114.001709. https://www.ahajournals.org/doi/pdf/10.1161/JAHA.114.001709.
38. ES Ford, UA Ajani, JB Croft, JA Critchley, DR Labarth, TE Kottke, WH Giles, S Capewell. Explaining the Decrease in U.S. Deaths from Coronary Disease, 1980–2000. N Engl J Med 2007; 356:2388–2398. DOI: 10.1056/NEJMsa053935. Cholesterol Treatment Trialists Collaboration (2010).